Authors Laurent Gatto, Johannes Rainer and Sebastian Gibb with contributions from Guangchuang Yu, Samuel Wieczorek, Vasile-Cosmin Lazar, Vladislav Petyuk, Thomas Naake, Richie Cotton and Martina Fisher.
Galaxy integration ABiMS TEAM - SU/CNRS - Station biologique de Roscoff and Yann Guitton - LABERCA Part of Workflow4Metabolomics.org [W4M]
Contact support@workflow4metabolomics.org for any questions or concerns about the Galaxy implementation of this tool.
MSnbase readMSData
Description
Reads as set of XML-based mass-spectrometry data files and generates an MSnExp object. This function uses the functionality provided by the ‘mzR’ package to access data and meta data in ‘mzData’, ‘mzXML’ and ‘mzML’.
Workflow position
Upstream tools
| Name | Format |
|---|---|
| Upload File | mzxml,mzml,mzdata,netcdf,zip |
The easier way to process is to create a Dataset Collection of the type List
Downstream tools
| Name | Output file | Format |
|---|---|---|
| xcms.findChromPeaks | *.raw.RData | rdata.msnbase.raw |
Example of a metabolomic workflow
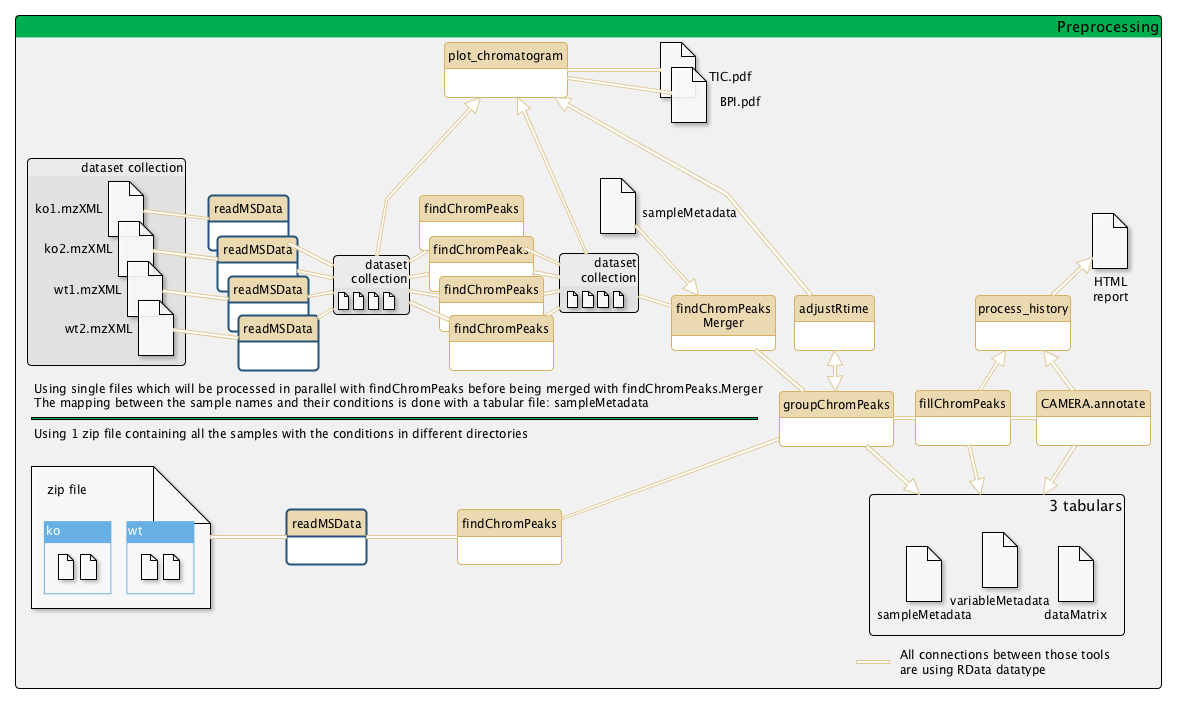
Input files
| Parameter : num + label | Format |
|---|---|
| OR : Zip file | zip |
| OR : Single file | mzXML, mzML, mzData, netCDF |
Choose your inputs
You have two methods for your inputs:
Single file (recommended): You can put a single file as input. That way, you will be able to launch several readMSData and findChromPeaks in parallel and use "findChromPeaks Merger" before groupChromPeaks.Zip file: You can put a zip file containing your inputs: myinputs.zip (containing all your conditions as sub-directories).
Zip file: Steps for creating the zip file
Step1: Creating your directory and hierarchize the subdirectories
VERY IMPORTANT: If you zip your files under Windows, you must use the 7Zip software, otherwise your zip will not be well unzipped on the W4M platform (corrupted zip bug).
Your zip should contain all your conditions as sub-directories. For example, two conditions (mutant and wild): arabidopsis/wild/01.raw arabidopsis/mutant/01.raw
Step2: Creating a zip file
Create your zip file (e.g. arabidopsis.zip).
Step 3 : Uploading it to our Galaxy server
Advices for converting your files into mzXML format (XCMS input)
We recommend you to convert your raw files into mzXML in centroid mode (smaller files); this way the files will be compatible with the xmcs centWave algorithm.
We recommend you the following parameters:
Use Filtering: True
Use Peak Picking: True
Peak Peaking -Apply to MS Levels: All Levels (1-) : Centroid Mode
Use zlib: 64
Binary Encoding: 64
m/z Encoding: 64
Intensity Encoding: 64
Output files
xset.RData: rdata.msnbase.raw format
Rdata file that is necessary in the second step of the workflow "xcms.findChromPeaks".
sampleMetadata.tsv (only when a zip is used)
Tabular file that contains for each sample its associated class and polarity (positive,negative and mixed).This file is necessary in further steps of the workflow, as the Anova and PCA steps for example.You get a sampleMetadata.tsv only if you use a zip. Otherwise, you have to provide one for the findChromPeaks Merger step.
Changelog/News
Version 2.16.1+galaxy0 - 08/04/2019
- UPGRADE: upgrade the MSnbase version from 2.8.2 to 2.16.1 (see MSnbase News). Almost all the new features may not concern our usage of MSnbase.
Version 2.8.2.1 - 30/04/2019
- BUGFIX: remove the pre-compute of the chromatograms which was memory consuming. Now, only xcms plot chromatogram will generate the Chromatograms.
Version 2.8.2.0 - 08/04/2019
- UPGRADE: upgrade the MSnbase version from 2.4.0 to 2.8.2 (see MSnbase News). Almost all the new features may not concern our usage of MSnbase.
Version 2.4.0.0 - 29/03/2018
- NEW: a new dedicated tool to read the raw data. This function was previously included in xcms.findChromPeaks. This way, you will now be able to display TICs and BPCs before xcms.findChromPeaks.