This tool searches MS/MS spectra against a database using X!Tandem.
For a complete set of parameters and their default values see the X!Tandem parameters documentation page . Parameters that are not made available in the UI above but are listed in the given link are submitted with their default values.
For more information on the refine step see: Why should I use "refinement" to find modifications? .
For more information on the expectation value calculation see: A Method for Assessing the Statistical Significance of Mass Spectrometry-Based Protein Identifications Using General Scoring Schemes , David Fenyö and Ronald C. Beavis, Anal. Chem., 2003, 75, 768-774. This reference describes how peptides are scored by X!Tandem. The expectation values on the individual peptides are calculated using this method.
Output
This tools returns the X!Tandem XML output which can be converted to MzIdentML using the DBSearch converter tool.
It also returns an HTML file with the list of peptides and the option to visualize the peptide to spectrum match using an embedded spectrum viewer.
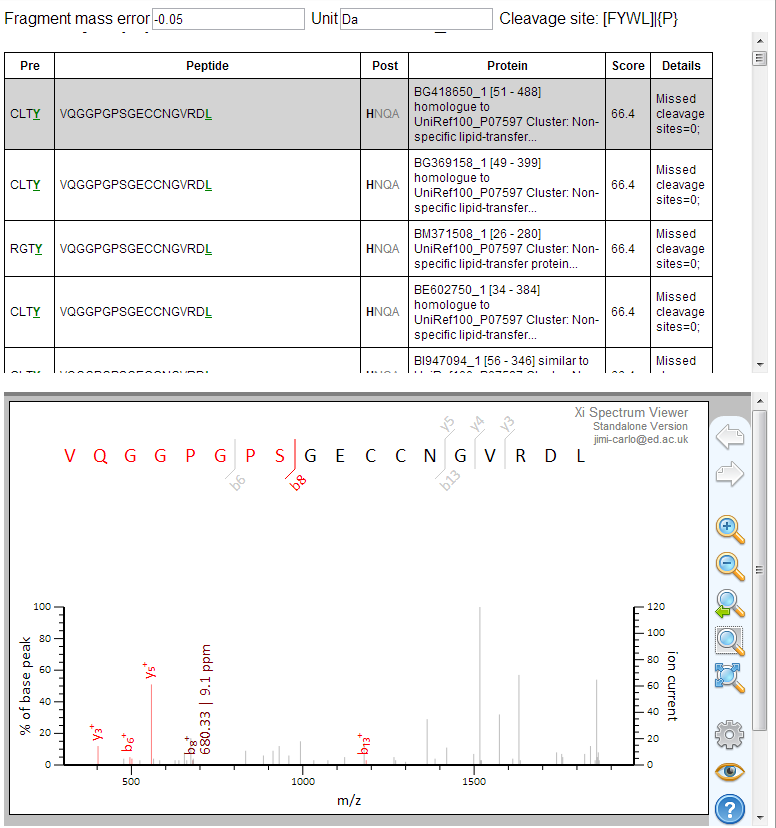
Last but not least, it returns the list of identifications in TSV (tab separated values) format for users that are satisfied with this and do not need further processing steps like protein inference.
For the GPM web UI of X!Tandem see: http://ppp.thegpm.org/tandem/thegpm_ppp.html