EncyclopeDIA Quantify
EncyclopeDIA is library search engine comprised of several algorithms for DIA data analysis and can search for peptides using either DDA-based spectrum libraries or DIA-based chromatogram libraries. See: https://bitbucket.org/searleb/encyclopedia/wiki/Home
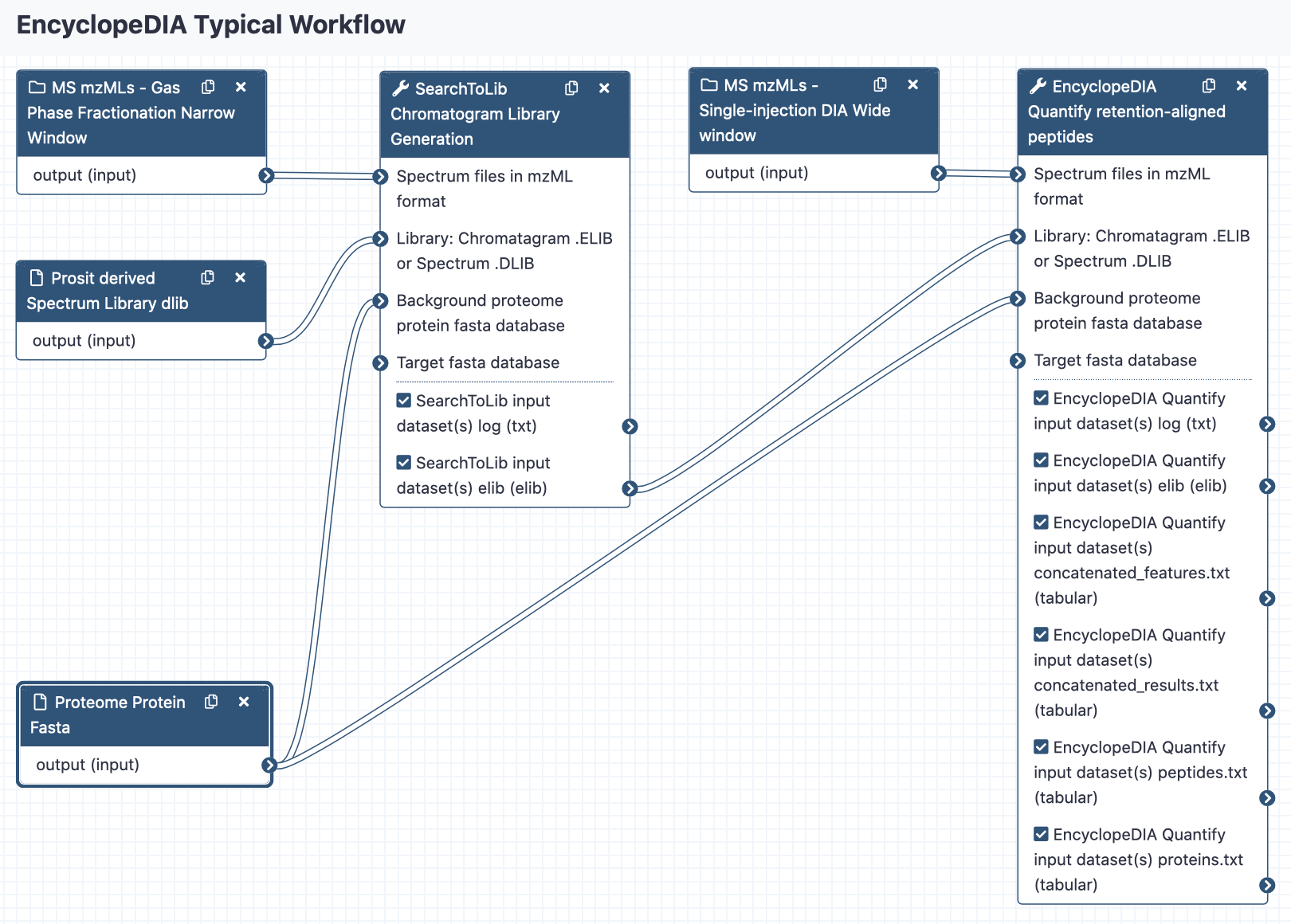
EncyclopeDIA Quantify retention-time aligns peptides from the chromatogram library and produces quantitation results.
Inputs
Spectrum files in mzML format
A chromatogram library that can be generated by SearchToLib
A protein data base in fasta format
The MSConvert command can be used to convert and deconvolute DIA raw files to mzML format. You need to use these options:
msconvert --zlib --64 --mzML --simAsSpectra --filter "peakPicking true 1-" --filter "demultiplex optimization=overlap_only" *.raw
Outputs
- A log file
- A Chromatogram Library (.elib)
- The identified features in tabular format Feature values of scans that are used by percolator to determine matches.
- The identified Peptide Spectral Match results in tabular format Columns: PSMId, score, q-value, posterior_error_prob, peptide, proteinIds
- The identified peptides in tabular format Per peptide: the normalized intensity for each scan file. Columns: Peptide, Protein, numFragments, intensity_in_file1, intensity_in_file2, ...
- The identified proteins in tabular format Per protein: the normalized intensity for each scan file. Columns: Protein, NumPeptides, PeptideSequences, intensity_in_file1, intensity_in_file2, ...
Typical DIA SearchToLib Workflow
Two sets of Mass Spec MS/MS DIA data are collected for the experiment. In addition to collecting wide-window DIA experiments on each quantitative replicate, a pool containing peptides from every condition is measured using several staggered narrow-window DIA experiments.
SearchToLib is first run with the pooled narrow-window mzML files to create a combined DIA elib chromatogram library. If a Spectral library argument is provided, for example from Prosit, SearchToLIB uses EncyclopeDIA to search each input spectrum mzML file. Otherwise, SearchToLIB uses Walnut, a FASTA database search engine for DIA data that uses PECAN-style scoring.
- Prosit generates a predicted spectrum library of fragmentation patterns and retention times for every +2H and +3H tryptic peptide in a FASTA database, with up to one missed cleavage.
EncyclopeDIA Quantify is then run on the wide-window quantitative replicate mzML files using that chromatogram library, with the align between files option, to produce quantification results.