Authors Colin A. Smith csmith@scripps.edu, Ralf Tautenhahn rtautenh@gmail.com, Steffen Neumann sneumann@ipb-halle.de, Paul Benton hpaul.benton08@imperial.ac.uk and Christopher Conley cjconley@ucdavis.edu
Galaxy integration ABiMS TEAM - SU/CNRS - Station biologique de Roscoff and Yann Guitton - LABERCA Part of Workflow4Metabolomics.org [W4M]
Contact support@workflow4metabolomics.org for any questions or concerns about the Galaxy implementation of this tool.
xcms findChromPeaks
Description
This tool is used for preprocessing data from multiple LC/MS files (NetCDF, mzXML and mzData formats) using the xcms R package. It extracts ions from each sample independently, and using a statistical model, peaks are filtered and integrated. A tutorial on how to perform xcms preprocessing is available as GTN (Galaxy Training Network).
Workflow position
Upstream tools
| Name | Output file | Format |
|---|---|---|
| MSnbase.readMSData | *.raw.RData | rdata.msnbase.raw |
Downstream tools
| Name | Output file | Format |
|---|---|---|
| xcms.findChromPeaks Merger (single) | *.raw.xset.RData | rdata.xcms.findchrompeaks |
| xcms.groupChromPeaks (zip) | *.raw.xset.RData | rdata.xcms.findchrompeaks |
Example of a metabolomic workflow
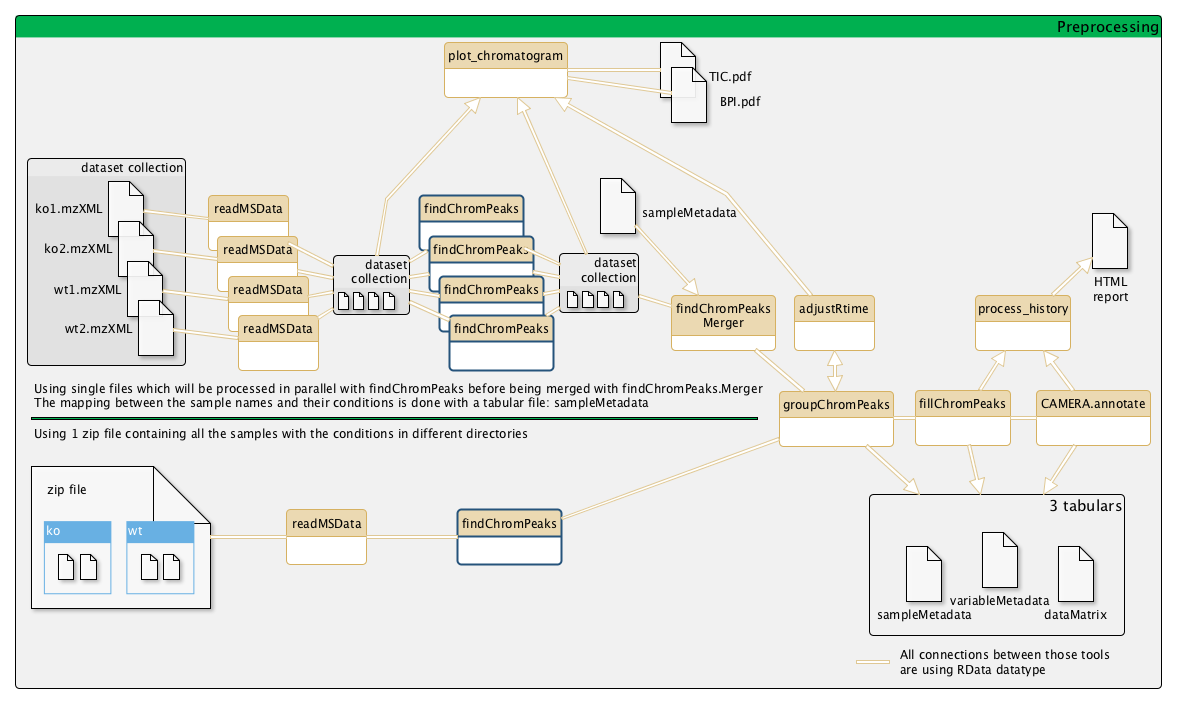
Parameters
Extraction method for peaks detection
MatchedFilter
The matchedFilter algorithm identifies peaks in the chromatographic time domain as described in [Smith 2006]. The intensity values are binned by cutting The LC/MS data into slices (bins) of a mass unit (‘binSize’ m/z) wide. Within each bin the maximal intensity is selected. The chromatographic peak detection is then performed in each bin by extending it based on the ‘steps’ parameter to generate slices comprising bins ‘current_bin - steps +1’ to ‘current_bin + steps - 1’. Each of these slices is then filtered with matched filtration using a second-derative Gaussian as the model peak shape. After filtration peaks are detected using a signal-to-ratio cut-off. For more details and illustrations see [Smith 2006].See the MatchedFilter_manual
CentWave
The centWave algorithm perform peak density and wavelet based chromatographic peak detection for high resolution LC/MS data in centroid mode [Tautenhahn 2008].Due to the fact that peak centroids are used, a binning step is not necessary.The method is capable of detecting close-by-peaks and also overlapping peaks. Some efforts are made to detect the exact peak boundaries to get precise peak integrals.See the CentWave_manual
CentWaveWithPredIsoROIs
This method performs a two-step centWave-based chromatographic peak detection: in a first centWave run peaks are identified for which then the location of their potential isotopes in the mz-retention time is predicted. A second centWave run is then performed on these regions of interest (ROIs). The final list of chromatographic peaks comprises all non-overlapping peaks from both centWave runs.See the CentWaveWithPredIsoROIs_manual
MSW
Wavelet based, used for direct infusion data. Continuous wavelet transform (CWT) can be used to locate chromatographic peaks on different scales.See the MSW_manual
For details and explanations concerning all the parameters and workflow of xcms package, see its manual and this example
Output files
xset.RData: rdata.xcms.findchrompeaks format
(single) RData files that are necessary in the second step of the workflow "xcms.groupChromPeaks" - must be merged first using "xcms.findChromPeaks Merger"(zip) RData file that is necessary in the second step of the workflow "xcms.groupChromPeaks".
Changelog/News
Version 3.12.0+galaxy* - 03/03/2020
- UPGRADE: upgrade the xcms version from 3.6.1 to 3.12.0 (see XCMS news)
Version 3.6.1+galaxy1 - 22/04/2020
- NEW: possibility to get a tabular file with all the chromatographic peaks obtained with the CentWave and MatchedFilter methods.
Version 3.6.1+galaxy* - 03/09/2019
- UPGRADE: upgrade the xcms version from 3.4.4 to 3.6.1 (see XCMS news)
Version 3.4.4.1 - 30/04/2019
- BUGFIX: remove the pre-compute of the chromatograms which was memory consuming. Now, only xcms plot chromatogram will generate the Chromatograms.
Version 3.4.4.0 - 08/02/2019
- UPGRADE: upgrade the xcms version from 3.0.0 to 3.4.4 (see XCMS news)
Version 3.0.0.0 - 08/03/2018
- UPGRADE: upgrade the xcms version from 1.46.0 to 3.0.0. So refactoring of a lot of underlying codes and methods. Some parameters may have been renamed.
- CHANGE: xcms.findChromPeaks no longer read the raw data. You have to run MSnbase readMSData first.
- NEW: a bunch of new options: Spectra Filters (previously scanrange), CentWave.(mzCenterFun, fitgauss, verboseColumns), MatchedFilter.(sigma, impute, baseValue, max), MSW.(verboseColumns), ...
- NEW: new Filters for Spectra
- NEW: new methods: CentWaveWithPredIsoROIs
- UPDATE: since xcms 3.0.0, some options are no more available: scanrange (replace by filters), profmethod, MatchedFilter.step, MatchedFilter.sigma, MSW.winSize.noise, MSW.SNR.method
- IMPROVEMENT: the advanced options are now in sections. It will allow you to access to all the parameters and to know their default values.
- IMPROVEMENT: the tool "should" be now more flexible in term of file naming: it "should" accept space and comma. But don't be too imaginative :)
- CHANGE: removing of the TIC and BPC plots. You can new use the dedicated tool "xcms plot chromatogram"
Version 2.1.1 - 29/11/2017
- BUGFIX: To avoid issues with accented letter in the parentFile tag of the mzXML files, we changed a hidden mechanim to LC_ALL=C
Version 2.1.0 - 22/02/2017
- NEW: The W4M tools will be able now to take as input a single file. It will allow to submit in parallel several files and merge them afterward using "xcms.xcmsSet Merger" before "xcms.group".
- BUGFIX: the default value of "matchedFilter" -> "Step size to use for profile generation" which was of 0.01 have been changed to fix with the XMCS default values to 0.1
Version 2.0.11 - 22/12/2016
- BUGFIX: propose scanrange for all methods
Version 2.0.10 - 22/12/2016
- BUGFIX: when having only one group (i.e. one folder of raw data) the BPC and TIC pdf files do not contain any graph
Version 2.0.9 - 06/07/2016
- UPGRADE: upgrade the xcms version from 1.44.0 to 1.46.0
Version 2.0.8 - 06/04/2016
- TEST: refactoring to pass planemo test using conda dependencies
Version 2.0.7 - 10/02/2016
- BUGFIX: better management of errors. Datasets remained green although the process failed
- BUGFIX/IMPROVEMENT: New checking steps around the imported data in order to raise explicte error message before or after launch XCMS: checking of bad characters in the filenames, checking of the XML integrity and checking of duplicates which can appear in the sample names during the XCMS process because of bad characters
- BUGFIX/IMPROVEMENT: New step to check and delete bad characters in the XML: accented characters in the storage path of the mass spectrometer
- UPDATE: refactoring of internal management of inputs/outputs
- TEST: refactoring to feed the new report tool
Version 2.0.2 - 18/01/2016
- BUGFIX: Some zip files were tag as "corrupt" by R. We have changed the extraction mode to deal with thoses cases.
Version 2.0.2 - 09/10/2015
- BUGFIX: Some users reported a bug in xcms.xcmsSet. The preprocessing stops itself and doesn't import the whole dataset contained in the zip file without warning. But meanwhile, please check your samplemetadata dataset and the number of rows.
Version 2.0.2 - 02/06/2015
- NEW: The W4M workflows will now take as input a zip file to ease the transfer and to improve dataset exchange between tools and users. (See How_to_upload). The previous "Library directory name" is still available but we invite user to switch on the new zip system as soon as possible.
- IMPROVEMENT: new datatype/dataset formats (rdata.xcms.raw, rdata.xcms.group, rdata.xcms.retcor ...) will facilitate the sequence of tools and so avoid incompatibility errors.
- IMPROVEMENT: parameter labels have changed to facilitate their reading.