Description
This tool implements dada2's assignTaxonomy and assignSpecies functions.
- assignTaxonomy assigns taxonomy to the sequence variants. The DADA2 package provides a native implementation of the naive Bayesian classifier method for this purpose (see Wang et al. 2007, kmer size 8 and 100 bootstrap replicates). The assignTaxonomy function takes as input a set of sequences to be classified and a training set of reference sequences with known taxonomy, and outputs taxonomic assignments with at least minBoot bootstrap confidence. Properly formatted reference files for several popular taxonomic databases are available http://benjjneb.github.io/dada2/training.html
- assignSpecies makes species level assignments based on exact matching between ASVs and sequenced reference strains. Recent analysis suggests that exact matching (or 100% identity) is the only appropriate way to assign species to 16S gene fragments. Currently, species-assignment training fastas are available for the Silva and RDP 16S databases.
Usage
Input
- A list of sequences contained in the results of removeBimeraDenovo or sequenceTable (note that also the results of dada, and mergePairs are accepted).
- Reference data bases for taxonomic and species/genus level assignment. Several cached data bases can be chosen (ask your Galaxy admin if they are missing). For using custom data bases see below.
Output
- A table containing the assigned taxonomies exceeding the minBoot level of bootstrapping confidence. Rows correspond to the provided sequences, columns to the taxonomic levels. NA indicates that the sequence was not consistently classified at that level at the minBoot threshold.
- Optionally two columns for the genus and species taxonomic levels can be added. NA indicates that the sequence was not classified at that level.
- If outputBootstraps checked, a table containing the assigned taxonomies (named "taxa") and the bootstrap values (named "boot") will be returned.
Overview
The intended use of the dada2 tools for paired sequencing data is shown in the following image.
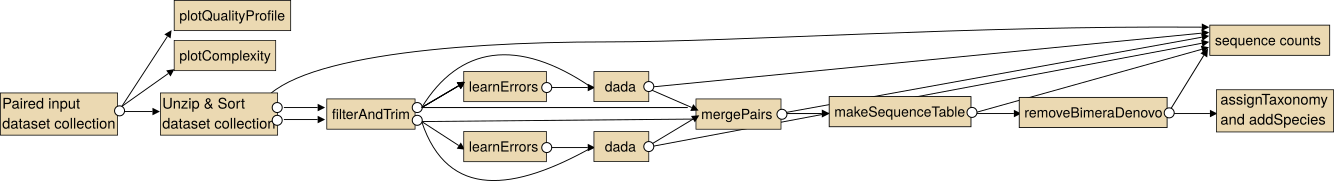
Note: In particular for the analysis of paired collections the collections should be sorted lexicographical before the analysis.
For single end data you the steps "Unzip collection" and "mergePairs" are not necessary.
More information may be found on the dada2 homepage:: https://benjjneb.github.io/dada2/index.html (in particular tutorials) or the documentation of dada2's R package https://bioconductor.org/packages/release/bioc/html/dada2.html (in particular the pdf which contains the full documentation of all parameters)
Custom Reference data sets
For ** taxonomy assignment ** the following is needed:
- a reference fasta data base
- a comma separated list of taxonomic ranks present in the reference data base
The reference fasta data base for taxonomic assignment (fasta or compressed fasta) needs to encode the taxonomy corresponding to each sequence in the fasta header lines in the following fashion (note, the second sequence is not assigned down to level 6):
>Level1;Level2;Level3;Level4;Level5;Level6; ACCTAGAAAGTCGTAGATCGAAGTTGAAGCATCGCCCGATGATCGTCTGAAGCTGTAGCATGAGTCGATTTTCACATTCAGGGATACCATAGGATAC >Level1;Level2;Level3;Level4;Level5; CGCTAGAAAGTCGTAGAAGGCTCGGAGGTTTGAAGCATCGCCCGATGGGATCTCGTTGCTGTAGCATGAGTACGGACATTCAGGGATCATAGGATAC
The list of required taxonomic ranks could be for instance: "Kingdom,Phylum,Class,Order,Family,Genus"
The reference data base for ** species assignment ** is a fasta file (or compressed fasta file), with the id line formatted as follows:
>ID Genus species ACCTAGAAAGTCGTAGATCGAAGTTGAAGCATCGCCCGATGATCGTCTGAAGCTGTAGCATGAGTCGATTTTCACATTCAGGGATACCATAGGATAC >ID Genus species CGCTAGAAAGTCGTAGAAGGCTCGGAGGTTTGAAGCATCGCCCGATGGGATCTCGTTGCTGTAGCATGAGTACGGACATTCAGGGATCATAGGATAC