What it does
Sailfish is a tool for transcript quantification from RNA-seq data. It requires a set of target transcripts (either from a reference or de-novo assembly) to quantify. All you need to run Sailfish is a fasta file containing your reference transcripts and a (set of) fasta/fastq file(s) containing your reads. Sailfish runs in two phases; indexing and quantification. The indexing step is independent of the reads, and only need to be run one for a particular set of reference transcripts and choice of k (the k-mer size). The quantification step, obviously, is specific to the set of RNA-seq reads and is thus run more frequently.
When the quantification output contains a number of columns: (1) Transcript ID, (2) Transcript Length, (3) Transcripts per Million (TPM) and (4) Estimated number of reads (an estimate of the number of reads drawn from this transcript given the transcript’s relative abundance and length).
The first two columns are self-explanatory, the next four are measures of transcript abundance and the final is a commonly used input for differential expression tools. The Transcripts per Million quantification number is computed as described in [1], and is meant as an estimate of the number of transcripts, per million observed transcripts, originating from each isoform. Its benefit over the F/RPKM measure is that it is independent of the mean expressed transcript length (i.e. if the mean expressed transcript length varies between samples, for example, this alone can affect differential analysis based on the K/RPKM.).
Fragment Library Types
There are numerous library preparation protocols for RNA-seq that result in sequencing reads with different characteristics. For example, reads can be single end (only one side of a fragment is recorded as a read) or paired-end (reads are generated from both ends of a fragment). Further, the sequencing reads themselves may be unstraned or strand-specific. Finally, paired-end protocols will have a specified relative orientation. To characterize the various different typs of sequencing libraries, we've created a miniature "language" that allows for the succinct description of the many different types of possible fragment libraries. For paired-end reads, the possible orientations, along with a graphical description of what they mean, are illustrated below:
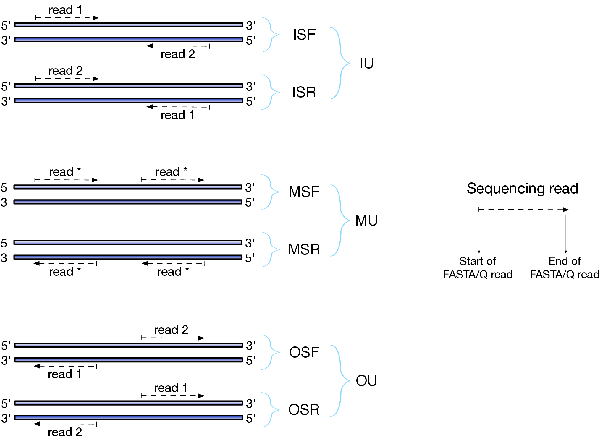
The library type string consists of three parts: the relative orientation of the reads, the strandedness of the library, and the directionality of the reads.
The first part of the library string (relative orientation) is only provided if the library is paired-end. The possible options are:
I = inward O = outward M = matching
The second part of the read library string specifies whether the protocol is stranded or unstranded; the options are:
S = stranded U = unstranded
If the protocol is unstranded, then we're done. The final part of the library string specifies the strand from which the read originates in a strand-specific protocol — it is only provided if the library is stranded (i.e. if the library format string is of the form S). The possible values are:
F = read 1 (or single-end read) comes from the forward strand R = read 1 (or single-end read) comes from the reverse strand
So, for example, if you wanted to specify a fragment library of strand-specific paired-end reads, oriented toward each other, where read 1 comes from the forward strand and read 2 comes from the reverse strand, you would specify -l ISF on the command line. This designates that the library being processed has the type "ISF" meaning, Inward (the relative orientation), Stranted (the protocol is strand-specific), Forward (read 1 comes from the forward strand).
The single end library strings are a bit simpler than their pair-end counter parts, since there is no relative orientation of which to speak. Thus, the only possible library format types for single-end reads are U (for unstranded), SF (for strand-specific reads coming from the forward strand) and SR (for strand-specific reads coming from the reverse strand).
A few more examples of some library format strings and their interpretations are:
IU (an unstranded paired-end library where the reads face each other)
SF (a stranded single-end protocol where the reads come from the forward strand)
OSR (a stranded paired-end protocol where the reads face away from each other,
read1 comes from reverse strand and read2 comes from the forward strand)
Note
Correspondence to TopHat library types
The popular TopHat RNA-seq read aligner has a different convention for specifying the format of the library. Below is a table that provides the corresponding sailfish/salmon library format string for each of the potential TopHat library types:
| TopHat | Salmon (and Sailfish) | |
|---|---|---|
| Paired-end | Single-end | |
| -fr-unstranded | -l IU | -l U |
| -fr-firststrand | -l ISR | -l SR |
| -fr-secondstrand | -l ISF | -l SF |
The remaining salmon library format strings are not directly expressible in terms of the TopHat library types, and so there is no direct mapping for them.