Authors Eric Fontanillas created the version 1 of this pipeline. Victor Mataigne developped version 2.
Galaxy integration Julie Baffard and ABiMS TEAM, Roscoff Marine Station
Contact support.abims@sb-roscoff.fr for any questions or concerns about the Galaxy implementation of this tool.Credits : Gildas le Corguillé, Misharl Monsoor
Description
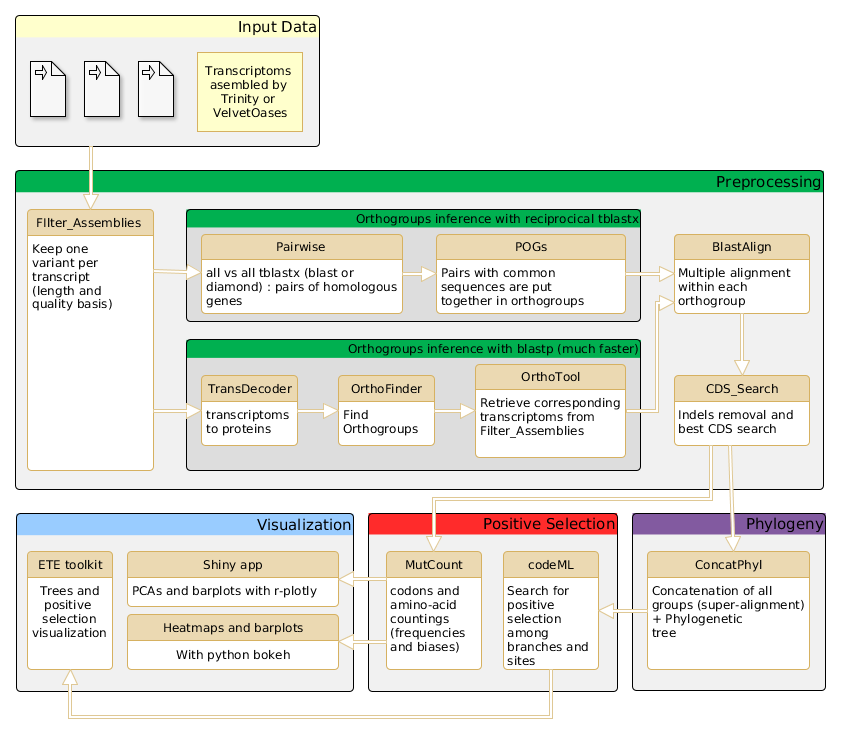
This tool takes files containing fasta sequences (from the CDS_Search in the AdaptSearch suite) and run RAxML to build a phylogeny.
full RAxML manual here
Parameters
- The choice of the format sequences is possible : proteic or nucleic
Several RAxML parameters can be set :
- Substitution model (-m) : Model of Binary (Morphological), Nucleotide, Multi-state, or Amino-Acid substitution
- Default : GTRGAMMA (nucleic), PROTCAT (proteic).
- Matrix : AA substitution model (when proteic inputs)
- Default : DAYHOFF
- random seed : Specifies a random number seed for the parsimony inferences. For all options/algorithms in RAxML that require some sort of randomization, this option must be specified. Make sure to pass different random number seeds to RAxML and not only 12345.
- Number of runs (-N) : Specifies the number of alternative runs.
- By default it's an integer of value 100.
- Use bootstopping criteria for number of runs :
- If selected, overxwrites the number of runs to use bootstopping criteria.
- Algorithm to execute (-f) : allows to choose what kind of algorithme RAxML shall execute.
- Default : Rapid bootsrap and best ML tree search (-f a).
- Multiple model assignement t oalignment partitions (-q) : an optional parameter. Permits to specify the file name which contains the assignment of models to alignment partitions for multiple models of substitution. For the syntax of this file please consult the manual.
- This option allows you to specify the regions of your alignment for which an individual model of nucleotide substitution should be estimated. This will typically be useful to infer trees for long multi-gene alignments.
- Rapid bootstrapping random seed (-x) : Specify an integer number (random seed) and turn on rapid bootstrapping.
- In addition to the best tree search. By default, this option is choosen.
Inputs
- Files from Filter Assemblies : a set of fasta files (one file per species), e.g. the outputs of the first tool of the AdaptSearch suite.
- Used to retrieve all the species names.
- Alignment files without indels : a set of fasta files with aligned sequences (with the same species than into the previous parameter), e.g the outputs of the CDS_Search tool of the AdaptSearch suite.
Outputs
This tool, produces the following files :
- Phylogeny :
- the general output. It gives the information about the concatenation (statistics) and the RAxML run.
- Phylogeny_concatenation_fasta_aa :
- contains the sequences concatenated in fasta format when you choose the option proteic.
- Phylogeny_concatenation_phylip_aa :
- contains the sequences concatenated in phylip format when you choose the option proteic.
- Phylogeny_concatenation_nexus_aa :
- contains the sequences concatenated in nexus format when you choose the option proteic.
- Phylogeny_concatenation_fasta_nuc :
- contains the sequences concatenated in fasta format when you choose the option nucleic.
- Phylogeny_concatenation_phylip_nuc :
- contains the sequences concatenated in phylip format when you choose the option nucleic. it's this output which is used for the RAxML run.
- Phylogeny_concatenation_nexus_nuc :
- contains the sequences concatenated in nexus format when you choose the option nucleic.
- Phylogeny_RAxML_BestTree** :
- the output of RAxML run which contains the Best Tree found.
- Phylogeny_RAxML_BiPartitionBranchLabel :
- the output of RAxML run which contains the Best Tree found with supported values as branch labels.
- Phylogeny_RAxML_BiPartition :
- the output of RAxML run which contains the Best Tree found with supported values.
- Phylogeny_RAxML_BootStrap :
- the output of RAxML run which contains all the boostrapped trees. The number of boostraped trees depending of the option -N (number of run).
The AdaptSearch Pipeline
Changelog
Version 2.0 - 06/07/2017
- NEW: Replace the zip between tools by Dataset Collection
Version 1.0 - 13/04/2017
- Add funtional test with planemo
- Planemo test with conda dependencies for raxml and python
- Scripts renamed + symlinks to the directory 'scripts'