Authors Colin A. Smith csmith@scripps.edu, Ralf Tautenhahn rtautenh@gmail.com, Steffen Neumann sneumann@ipb-halle.de, Paul Benton hpaul.benton08@imperial.ac.uk and Christopher Conley cjconley@ucdavis.edu
Galaxy integration ABiMS TEAM - UPMC/CNRS - Station biologique de Roscoff and Yann Guitton yann.guitton@oniris-nantes.fr - part of Workflow4Metabolomics.org [W4M]
Contact support@workflow4metabolomics.org for any questions or concerns about the Galaxy implementation of this tool.
Xcms.combinexsAnnos
Description
What it does?
This function check annotations of ion species with the help of a sample from opposite ion mode. As first step it searches for pseudospectra from the positive and the negative sample within a reten- tion time window. For every result the m/z differences between both samples are matched against specific rules, which are combinations from pos. and neg. ion species. As example M+H and M-H with a m/z difference of 2.014552. If two ions matches such a difference, the ion annotations are changed (previous annotation is wrong), confirmed or added. Returns the peaklist from one ion mode with recalculated annotations.
Details
Both xsAnnotate object should be full processed (grouping and annotation). Without previous anno- tation the resulting peaklist only includes annotation with matches peaks from both mode according to the rule(s). With ruleset=NULL the function only looks for M+H/M-H pairs. The ruleset is a two column matrix with includes rule indices from the rule table of both xsAnnotate objects. A ruleset (1,1) would create the M+H/M-H rule, since the first rule of xsa.pos@ruleset and xsa.neg@ruleset is M+H respectively M-H. Only rules with identical charge can be combined!
Workflow position
Upstream tools
| Name | Output file | Format | Parameter |
|---|---|---|---|
| xcms.annotatediffreport | xset.annotate_POS.RData | rdata.camera.positive | RData file |
| xcms.annotatediffreport | xset.annotate_NEG.RData | rdata.camera.positive | RData file |
Downstream tools
| Name | Output file | Format |
|---|---|---|
| Batch_correction | xset.combinexsAnnos.variableMetadata.tsv | Tabular |
| Filters | xset.combinexsAnnos.variableMetadata.tsv | Tabular |
| Univariate | xset.combinexsAnnos.variableMetadata.tsv | Tabular |
| Multivariate | xset.combinexsAnnos.variableMetadata.tsv | Tabular |
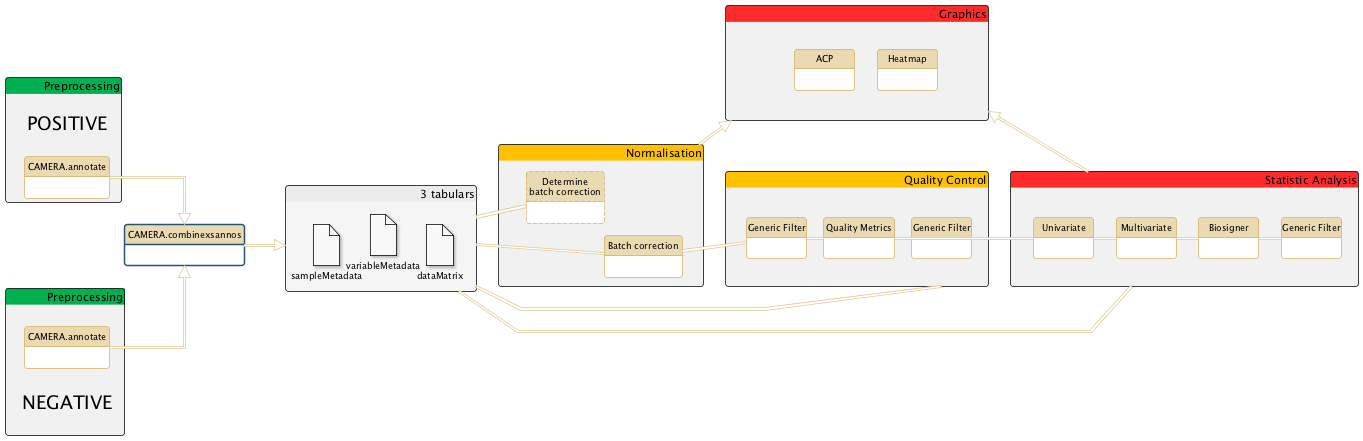
- The output file xset.annotateDiffreport.variableMetadata.tsv is a tabular file. You can continue your analysis using it in the following tools:
- Batch_correctionFiltersUnivariateMultivariate PCA, PLS and OPLS
Place of CAMERA.combinexsannot after XCMS part of the metabolomic workflow
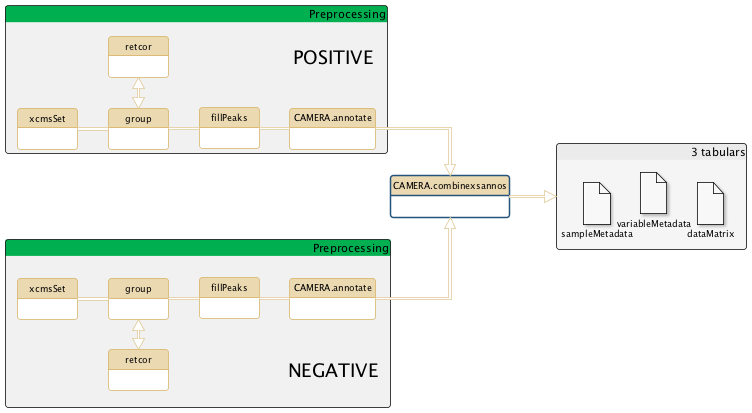
General schema of the metabolomic workflow
Input files
| Parameter : label | Format |
|---|---|
| Positive RData ion mode | rdata.camera.positive |
| Negative RData ion mode | rdata.camera.negative |
Output files
xset.combinexsAnnos.variableMetadata.tsv
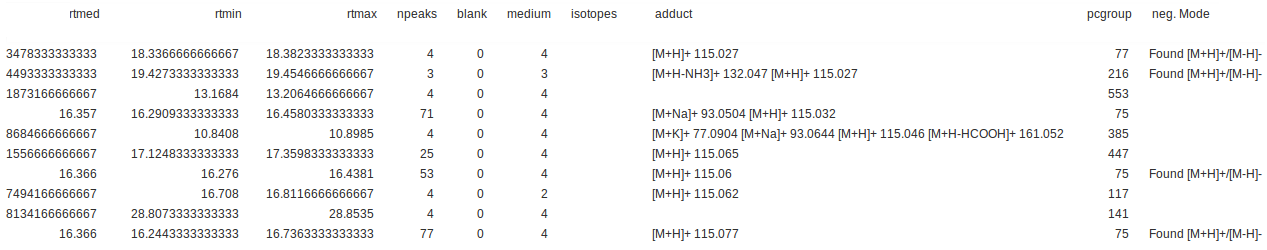
A tabular file which is similar to the diffreport result , within additional columns containing the annotation results.For each metabolite (row) :the value of the intensity in each sample, fold, tstat, pvalue, anova, mzmed, mzmin, mzmax, rtmed, rtmin, rtmax, npeaks, isotopes, adduct, pcgroup and neg (or pos). Mode
xset.combinexsAnnos.Rdata
Rdata file, that be used outside Galaxy in R.
Working example
Input files
Positive RData ion mode -> POS.xset.annotateDiffreport.RDataNegative RData ion mode -> NEG.xset.annotateDiffreport.RData
Parameters
pos -> positivetol -> 2 (default)ruleset -> 1,1 (default)
Output files
Example of an xset.combinexsAnnos.variableMetadata.tsv output:
Changelog/News
Version 2.2.2 - 01/03/2018
- UPGRADE: upgrate the CAMERA version from 1.26.0 to 1.32.0
Version 2.0.7 - 29/11/2017
- BUGFIX: To avoid issues with accented letter in the parentFile tag of the mzXML files, we changed a hidden mechanim to LC_ALL=C
Version 2.0.6 - 10/02/2017
- IMPROVEMENT: Synchronize the variableMetadata export option with the other tools (xcms.group, xcms.fillpeaks, camera.annotate)
Version 2.0.5 - 22/12/2016
- IMPROVEMENT: add the possibility to add a personal Matrix of matching rules (ruleset)
Version 2.0.4 - 21/04/2016
- UPGRADE: upgrate the CAMERA version from 1.22.0 to 1.26.0
Version 2.0.3 - 10/02/2016
- BUGFIX: better management of errors. Datasets remained green although the process failed
- UPDATE: refactoring of internal management of inputs/outputs
Version 2.0.1 - 07/06/2015
- IMPROVEMENT: new datatype/dataset formats (rdata.camera.positive, rdata.camera.negative, rdata.camera.quick ...) will facilitate the sequence of tools and so avoid incompatibility errors.
- IMPROVEMENT: parameter labels have changed to facilitate their reading.
Version 2.0.0 - 09/06/2015
- NEW: combinexsAnnos Check CAMERA ion species annotation due to matching with opposite ion mode