Description
Filters and trims a FASTQ dataset (can be compressed) based on several user-definable criteria, and outputs a compressed FASTQ data set containing those trimmed reads which passed the filters. For paired end data forward and reverse FASTQ datasets can be provided as pair of FASTQ datasets (or two separate data sets), in which case filtering is performed on the forward and reverse reads independently, and both reads must pass for the read pair to be in the output.
Usage
Input is a FASTQ dataset (or a pair in case of paired end data) containing all reads of a sample. It is suggested to organize them in a (paired) collection (in particular if you have multiple samples).
Output is a (paired) collection of filtered and trimmed paired FASTQ datasets (again one data set or pair per sample).
Upstream dada2 tools are dada2: learnErrorRates and dada2: dada. Note that these tools do not work on paired end data. So, if you have paired end data you need to split the generated paired collection into one containing the forward reads and one containing the reverse reads. This can be done by the unzip collection tool.
An additional tabular output gives the number of reads before and after trimming. This can data set can be used as input for dada2: sequence counts to track the sequence counts for each sample through all dada2 pipeline step.
Details
Trimming and filtering:
- Truncation of the read length is enforced after trimming of the right end.
- The long read filter is applied before trimming and the short read filter after trimming.
- For details on the calculation of the number of expected errors see also https://doi.org/10.1093/bioinformatics/btv401
String present at the start of valid reads (orient.fwd):
This string is compared to the start of each read, and the reverse complement of each read. If it exactly matches the start of the read, the read is kept. If it exactly matches the start of the reverse complement read, the read is reverse-complemented and kept. Otherwise the read if filtered out. For paired reads, the string is compared to the start of the forward and reverse reads, and if it matches the start of the reverse read the reads are swapped and kept. The primary use of this parameter is to unify the orientation of amplicon sequencing libraries that are a mixture of forward and reverse orientations, and that include the forward primer on the reads.
Low complexity filter kmer threshold"
If greater than 0, reads with an effective number of kmers less than this value will be removed. The effective number of kmers is determined as a Shannon information approximation. The default kmer-size is 2, and therefore perfectly random sequences will approachan effective kmer number of 16 = 4 (nucleotides) ^ 2 (kmer size).
Notes
This step may be replaced by alternative tools to filter and trim short read data if the following is ensured:
- For paired end data unpaired reads must be removed.
- There must not be a read containing a non-canonical nucleotide (N).
Overview
The intended use of the dada2 tools for paired sequencing data is shown in the following image.
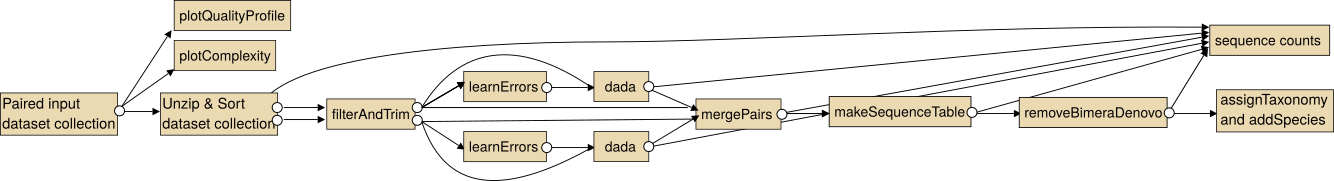
Note: In particular for the analysis of paired collections the collections should be sorted lexicographical before the analysis.
For single end data you the steps "Unzip collection" and "mergePairs" are not necessary.
More information may be found on the dada2 homepage:: https://benjjneb.github.io/dada2/index.html (in particular tutorials) or the documentation of dada2's R package https://bioconductor.org/packages/release/bioc/html/dada2.html (in particular the pdf which contains the full documentation of all parameters)