What it does
FROGS Pre-process filters and dereplicates amplicons for use in diversity analysis.
Inputs/Outputs
Inputs
Sample files added one after another or provide in an archive file (tar.gz).
Illumina inputs
Usage: For samples sequenced in paired-end. The amplicon length must be inferior to the length of the R1 plus R2 length. R1 and R2 are merged by the common region. Files: One R1 and R2 by sample (format FASTQ) Example: splA_R1.fastq.gz, splA_R2.fastq.gz, splB_R1.fastq.gz, splB_R2.fastq.gz
OR
Usage: For samples sequenced in single-ends or when R1 and R2 reads are already merged. Files: One sequence file by sample (format FASTQ). Example: splA.fastq.gz, splB.fastq.gz
454 inputs
Files: One sequence file by sample (format FASTQ) Example: splA.fastq.gz, splB.fastq.gz
Remark: In an archive if you use R1 and R2 files they names must end with _R1 and _R2. To upload an archive, see the "Upload archive" tool or if possible create symbolic link on your Galaxy account.
Outputs
Sequence file (dereplicated.fasta):
Only one file with all samples sequences (format FASTA). These sequences are dereplicated: strictly identical sequence are represented only one and the initial count is kept in count file.
Count file (count.tsv):
This file contains the count of all unique sequences in each sample (format TSV).
Summary file (report.html):
This file reports the number of remaining sequences after each filter (format HTML).
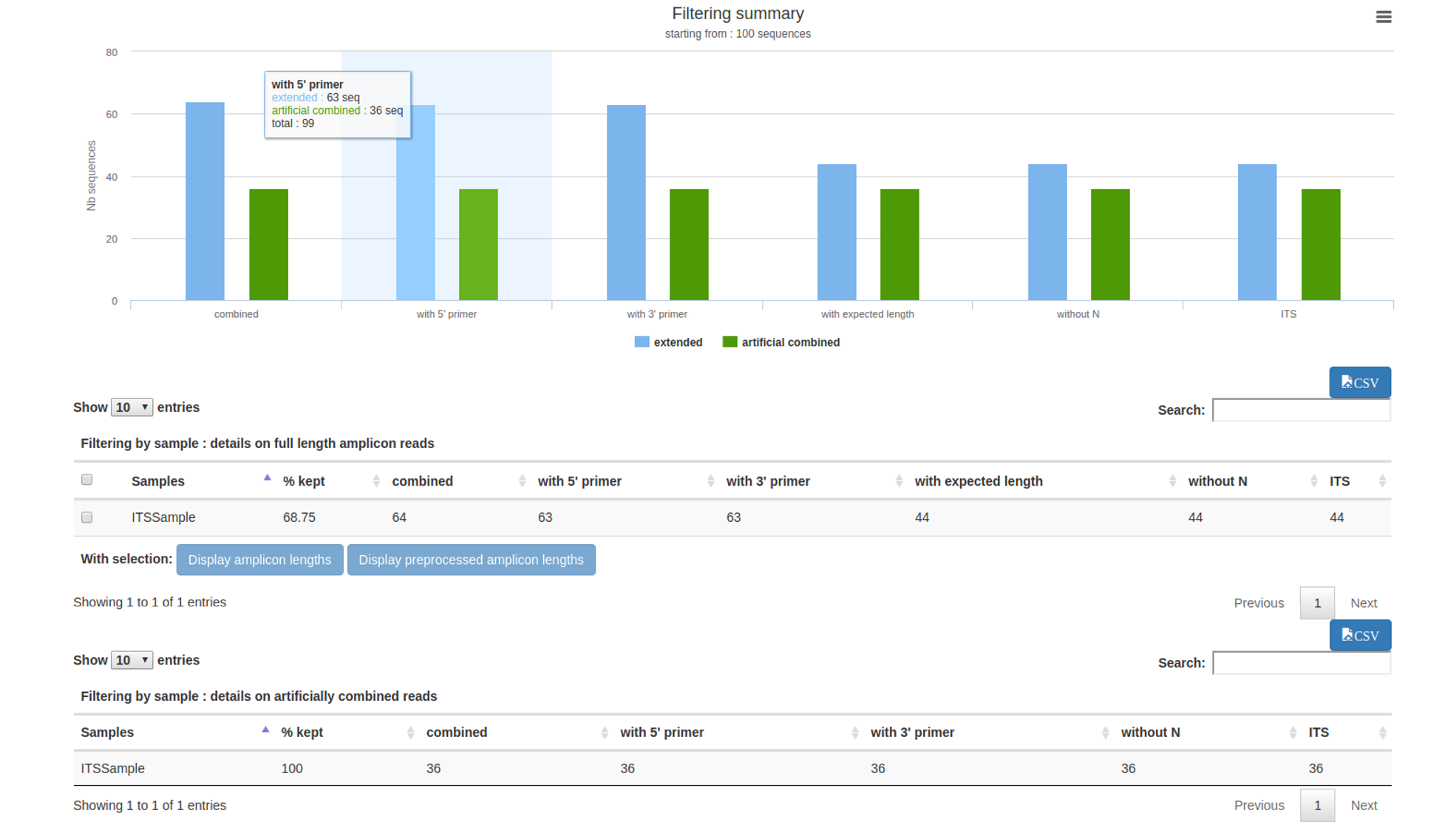
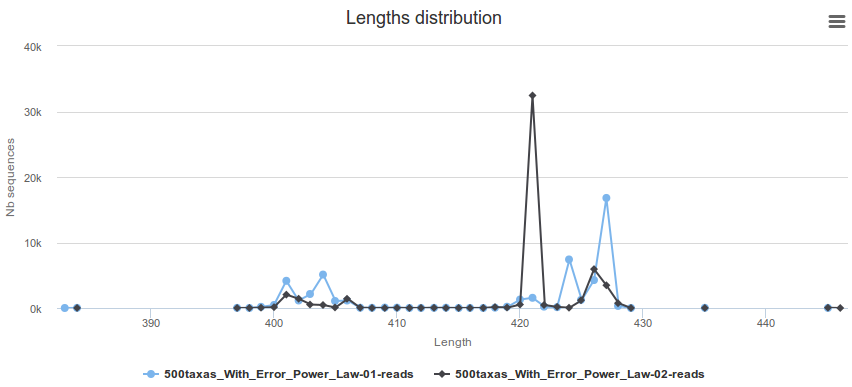
It also presents the length distribution of the remaining sequences.
How it works
| Steps | Illumina | 454 |
|---|---|---|
| 1 | For un-merged data: merges R1 and R2 with a maximum of M% mismatch in the overlaped region (FLASh). By default M is set to 10% | / |
| 2 | Filters merged sequences on their length which must be range between 'Minimum amplicon size' and 'Maximum amplicon size' | / |
| 3 | If sequencing protocol is the illumina standard protocol : Removes sequences where the two primers are not present and then remove primers in the remaining sequence (cutadapt). The primer search accepts 10% of differences | Removes sequences where the two primers are not present, removes primers sequence and reverse complement the sequences on strand - (cutadapt). The primer search accepts 10% of differences |
| 4 | Filters sequences on their length and with ambiguous nucleotides | the tool removes sequences with at least one homopolymer with more than seven nucleotides and with a distance of less than or equal to 10 nucleo-tides between two poor quality positions, i.e. with a Phred quality score lesser than 10 |
| 5 | Dereplicates sequences | Dereplicates sequences |
Advices/details on parameters
Primers parameters
The (Kozich et al. 2013 ) protocol uses custom sequencing primers which are also the PCR primers. In this case the reads do not contain the PCR primers.
In case of Illumina standard protocol, the primers must be provided in 5' to 3' orientation.
Example:
5' ATGCCC GTCGTCGTAAAATGC ATTTCAG 3'
Value for parameter 5' primer: ATGCC
Value for parameter 3' primer: ATTTCAG
Amplicons sizes parameters
The two following images show two examples of perfect values fors sizes parameters.
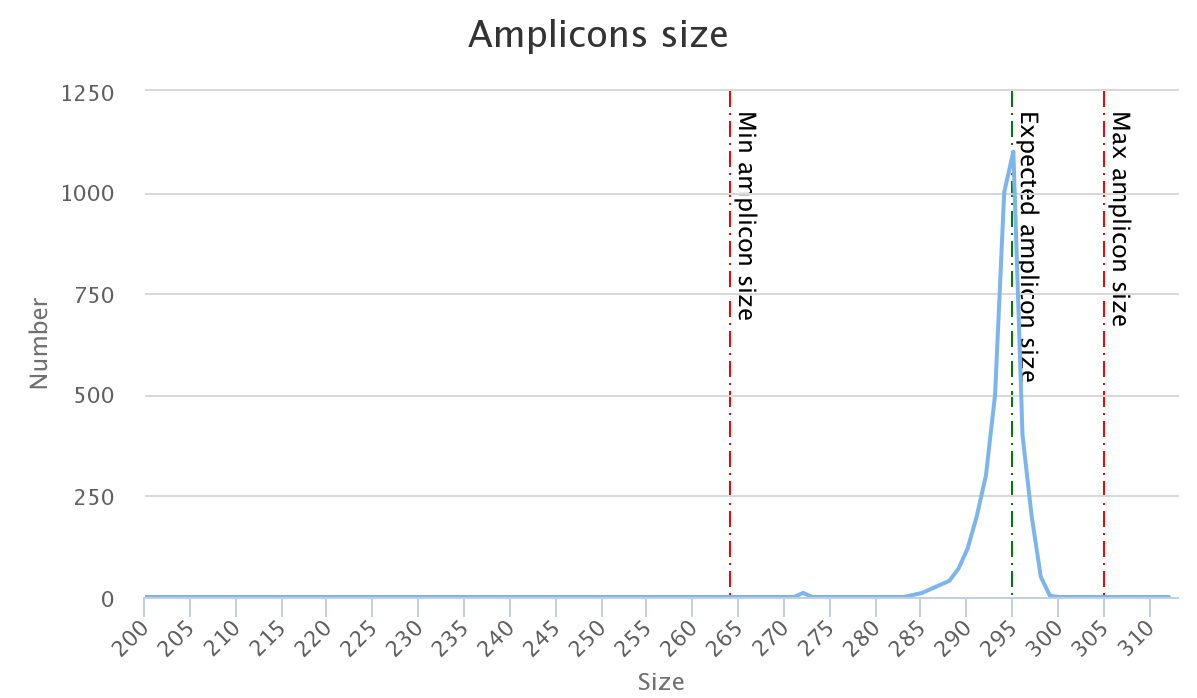
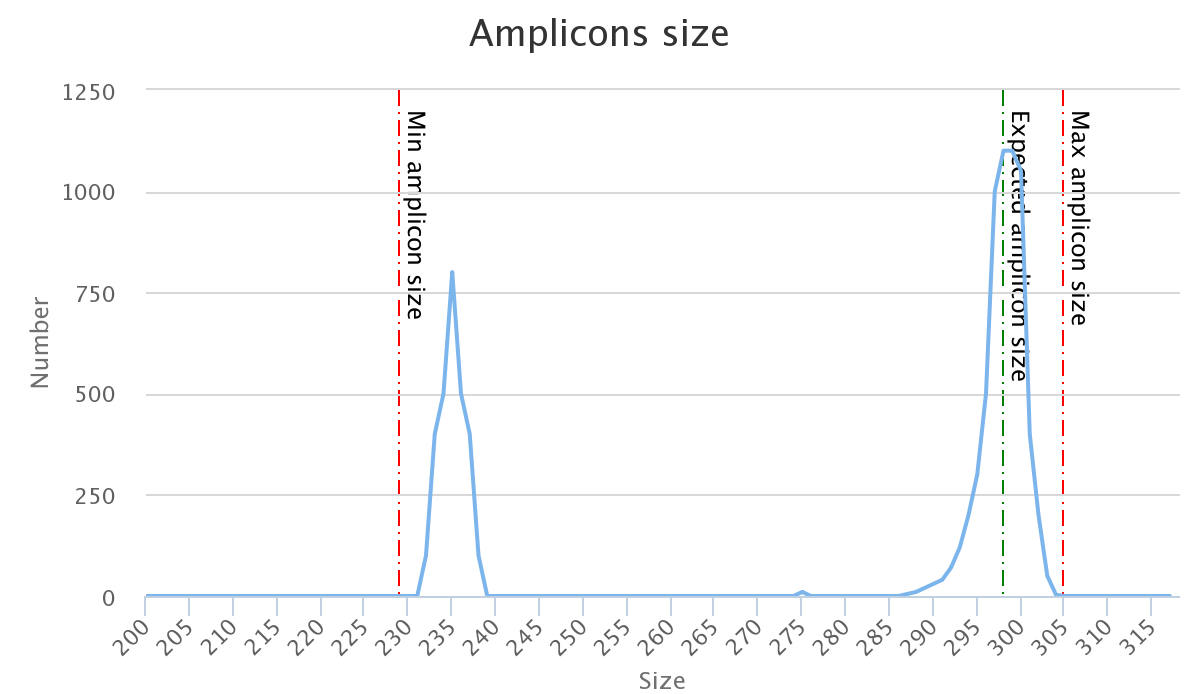
Don't worry the "Expected amplicon size" does not need to be very accurate.
If the filter 'overlapped' reduce drasticaly the number of sequences:
In un-merged Illumina data, the reduction of dataset by the overlapped filter is classicaly inferior than 20%. A loss of more than 20% in all samples can highlight a quality problem.
If the overlap between R1 and R2 is superior to 50 nucleotides and the quality of the end of the sequences is poor (see FastQC) you can try to cut the end of your sequences and relaunch the preprocess tool. You can either raise the mismatch percent in the overlapped region, but not too much!
Contact
Contacts: frogs@inra.fr
Repository: https://github.com/geraldinepascal/FROGS
Please cite the FROGS Publication: Frederic Escudie, Lucas Auer, Maria Bernard, Mahendra Mariadassou, Laurent Cauquil, Katia Vidal, Sarah Maman, Guillermina Hernandez-Raquet, Sylvie Combes, Geraldine Pascal; FROGS: Find, Rapidly, OTUs with Galaxy Solution, Bioinformatics, , btx791, https://doi.org/10.1093/bioinformatics/btx791
Depending on the help provided you can cite us in acknowledgements, references or both.