Description
The dada function takes as input amplicon sequencing reads and returns the inferred composition of the sample (or samples). Put another way, dada removes all sequencing errors to reveal the members of the sequenced community.
Usage
Input:
- A number of fastq(.gz) files (given as collection or multiple data sets)
- An dada2_errorrates data set computed with learnErrors
You can decide to compute the data jointly or in batches.
- Jointly (Process "samples in batches"=no): A single Galaxy job is started that processes all fastq data sets jointly. You may chose different pooling strategies: if the started dada job processes the samples individually, pooled, or pseudo pooled.
- In batches (Process "samples in batches"=yes): A separate Galaxy job is started for earch fastq data set. This is equivalent to joint processing and choosing to process samples individually.
While the single dada job (in case of joint processing) can use multiple cores on one compute node, batched processing distributes the work on a number of jobs (equal to the number of input fastq data sets) where each can use multiple cores. Hence, if you intend to or need to process the data sets individually, batched processing is more efficient -- in particular if Galaxy has access to a larger number of compute resources.
A typical use case of individual processing of the samples are large data sets for which the pooled strategy needs to much time or memory. Pseudo-pooling is recommended for those interested in detecting singleton ASVs in their samples
Output: a data set of type dada2_dada (which is a RData file containing the output of dada2's dada function).
The output of this tool can serve as input for dada2: mergePairs, dada2: removeBimeraDinovo, and "dada2: makeSequenceTable"
Details
Briefly, dada implements a statistical test for the notion that a specific sequence was seen too many times to have been caused by amplicon errors from currently inferred sample sequences. Overly abundant sequences are used as the seeds of new partitions of sequencing reads, and the final set of partitions is taken to represent the denoised composition of the sample. A more detailed explanation of the algorithm is found in the dada2 puplication (see below) and https://doi.org/10.1186/1471-2105-13-283. dada depends on a parametric error model of substitutions. Thus the quality of its sample inference is affected by the accuracy of the estimated error rates. All comparisons between sequences performed by dada depend on pairwise alignments. This step is the most computationally intensive part of the algorithm, and two alignment heuristics have been implemented in dada for speed: A kmer-distance screen and banded Needleman-Wunsch alignmemt.
Overview
The intended use of the dada2 tools for paired sequencing data is shown in the following image.
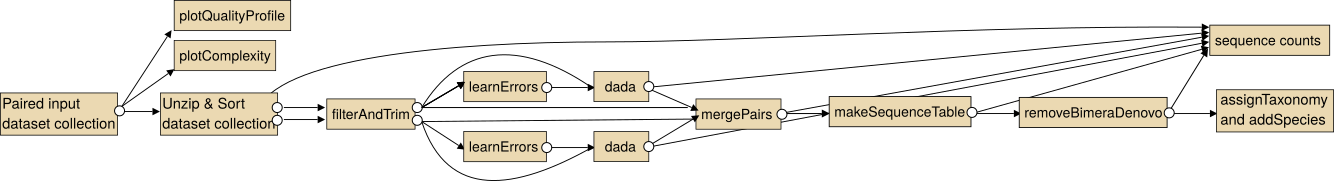
Note: In particular for the analysis of paired collections the collections should be sorted lexicographical before the analysis.
For single end data you the steps "Unzip collection" and "mergePairs" are not necessary.
More information may be found on the dada2 homepage:: https://benjjneb.github.io/dada2/index.html (in particular tutorials) or the documentation of dada2's R package https://bioconductor.org/packages/release/bioc/html/dada2.html (in particular the pdf which contains the full documentation of all parameters)