What It Does
Disclaimer Disclaimer this tool needs a peptide database and peptide spectrum matches.
An established method to identify acquired MS/MS spectra is the comparison of each spectrum with peptides in a reference database.
To measure the similarity of in silico generated spectra and measured MS/MS scans we use our own implementations of three established search enginge scores: SEQUEST, Andromeda and XTandem. Additionally, we also record quality control parameters such as the mass difference between the precursor ion and the theoretically calulated mass or the uniquness of each score in comparison to 'competing' peptides within the search space. The PSMStatistics tool utilizes semi supervised machine learning techniques to integrate search engine scores as well as the mentioned quality scores into one single consensus score.
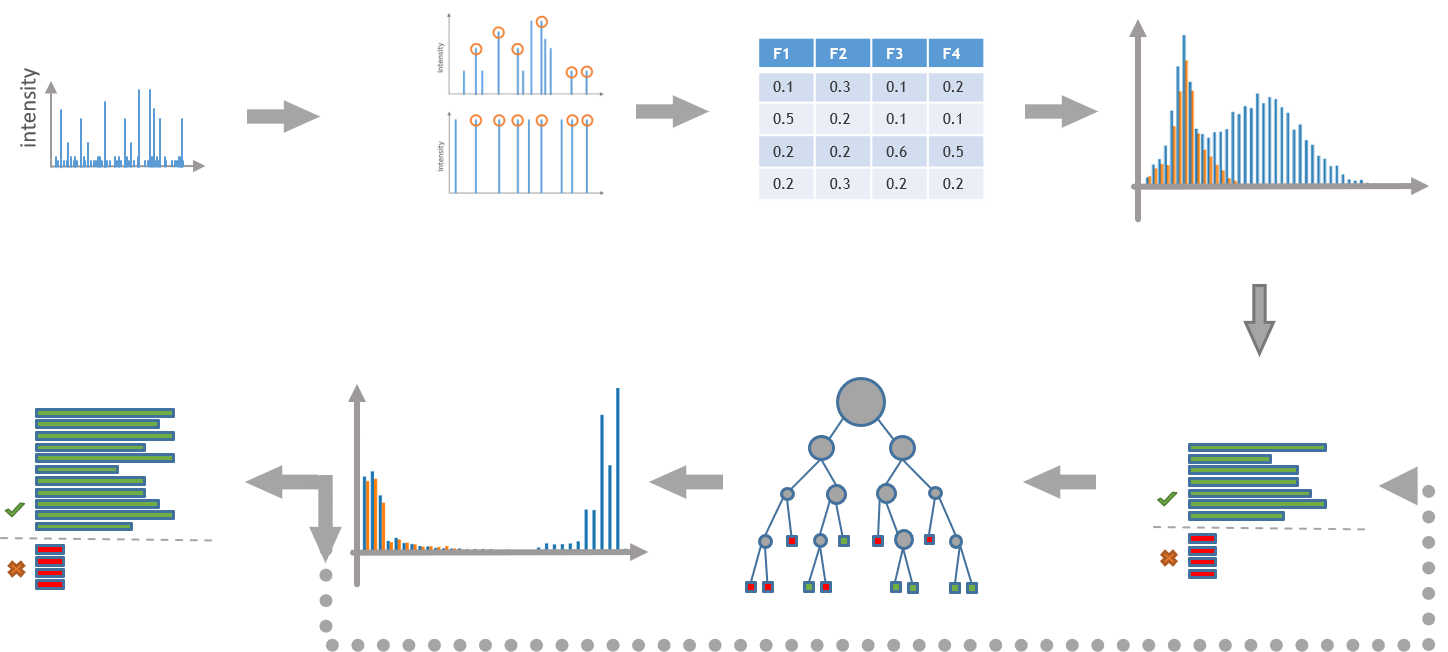
Since the search space is extended by so called decoys - reversed counterparts of peptides within the search space - we can estimate the distribution of 'true negatives' and calculate local (PEP values) and global (Q values) false discovery rates at each consensus score. The reported peptides at user defined local and global FDR cutoffs can then be used as inputs for any downstream analysis be it ProteinInference or PSMBasedQuantification.
Further Reading
Additional information about the tool can be found in the documentation.