What it does
Similar to merge, cluster report each set of overlapping or “book-ended” features in an interval file. In contrast to merge, cluster does not flatten the cluster of intervals into a new meta-interval; instead, it assigns an unique cluster ID to each record in each cluster. This is useful for having fine control over how sets of overlapping intervals in a single interval file are combined.
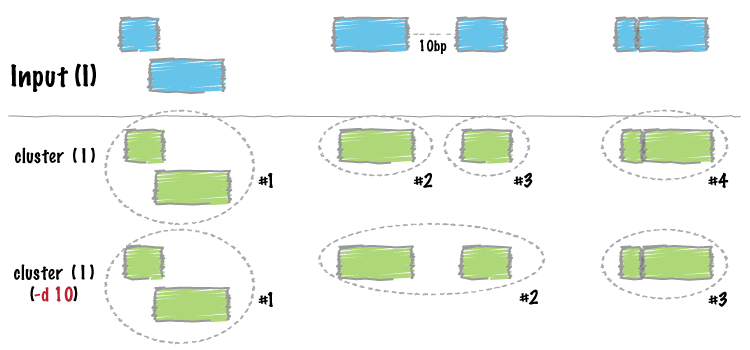
bedtools cluster requires that you presort your data by chromosome and then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed for BED files).
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper