What it does
This tool calculates the genome-wide coverage of intervals defined in a BAM or BED file and reports them in BedGraph format.
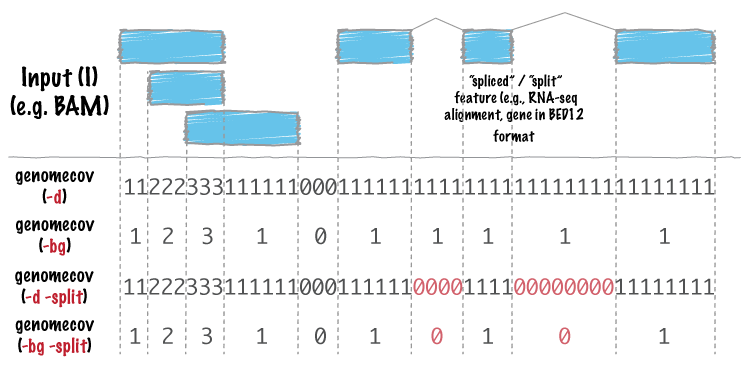
The input BED or BAM file must be sorted by chromosome name (but doesn't necessarily have to be sorted by start position).
Example 1
Input (BED format)- Overlapping, un-sorted intervals:
chr1 140 176 chr1 100 130 chr1 120 147
Output (BedGraph format)- Sorted, non-overlapping intervals, with coverage value on the 4th column:
chr1 100 120 1 chr1 120 130 2 chr1 130 140 1 chr1 140 147 2 chr1 147 176 1
Example 2 - with ZERO-Regions selected (assuming hg19)
Input (BED format)- Overlapping, un-sorted intervals:
chr1 140 176 chr1 100 130 chr1 120 147
BedGraph output will contain five columns:
- Chromosome name (or 'genome' for whole-genome coverage)
- Coverage depth
- The number of bases on chromosome (or genome) with depth equal to column 2.
- The size of chromosome (or entire genome) in base pairs
- The fraction of bases on chromosome (or entire genome) with depth equal to column 2.
Example Output:
chr2L 0 1379895 23011544 0.0599653 chr2L 1 837250 23011544 0.0363839 chr2L 2 904442 23011544 0.0393038 chr2L 3 913723 23011544 0.0397072 chr2L 4 952166 23011544 0.0413778 chr2L 5 967763 23011544 0.0420555 chr2L 6 986331 23011544 0.0428624 chr2L 7 998244 23011544 0.0433801 chr2L 8 995791 23011544 0.0432735 chr2L 9 996398 23011544 0.0432999
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper