What it does
By far, the most common question asked of two sets of genomic features is whether or not any of the features in the two sets “overlap” with one another. This is known as feature intersection. bedtools intersect allows one to screen for overlaps between two sets of genomic features. Moreover, it allows one to have fine control as to how the intersections are reported. bedtools intersect works with both BED/bedGraph/GFF/VCF/EncodePeak and BAM files as input.
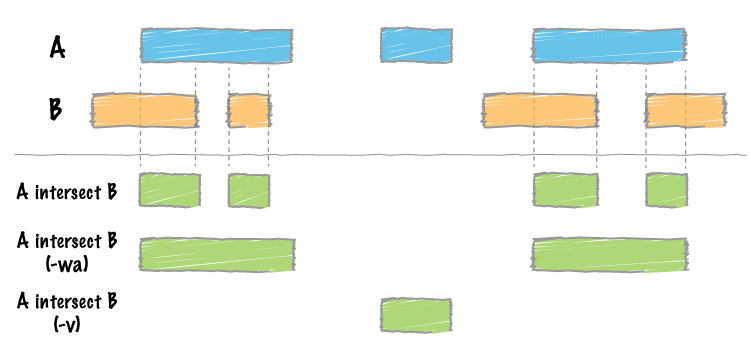
Note that each BAM alignment is treated individually. Therefore, if one end of a paired-end alignment overlaps an interval in the BED file, yet the other end does not, the output file will only include the overlapping end.
Note that a BAM alignment will be sent to the output file once even if it overlaps more than one interval in the BED file.
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper