What it does
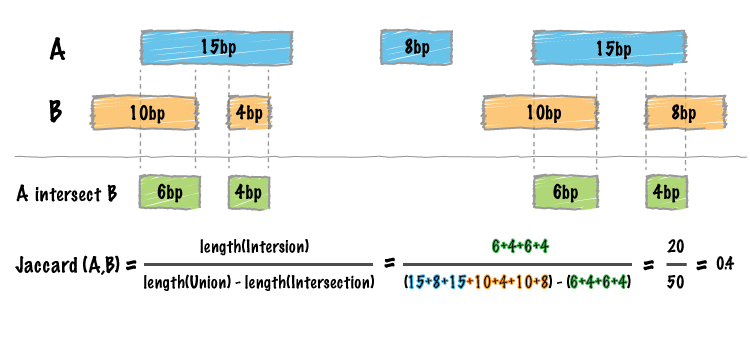
By default, bedtools jaccard reports the length of the intersection, the length of the union (minus the intersection), the final Jaccard statistic reflecting the similarity of the two sets, as well as the number of intersections. Whereas the bedtools intersect tool enumerates each an every intersection between two sets of genomic intervals, one often needs a single statistic reflecting the similarity of the two sets based on the intersections between them. The Jaccard statistic is used in set theory to represent the ratio of the intersection of two sets to the union of the two sets. Similarly, Favorov et al [1] reported the use of the Jaccard statistic for genome intervals: specifically, it measures the ratio of the number of intersecting base pairs between two sets to the number of base pairs in the union of the two sets. The bedtools jaccard tool implements this statistic, yet modifies the statistic such that the length of the intersection is subtracted from the length of the union. As a result, the final statistic ranges from 0.0 to 1.0, where 0.0 represents no overlap and 1.0 represent complete overlap.
The jaccard tool requires that your data is pre-sorted by chromosome and then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed for BED files).
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper