What it does
bedtools map allows one to map overlapping features in a B file onto features in an A file and apply statistics and/or summary operations on those features.
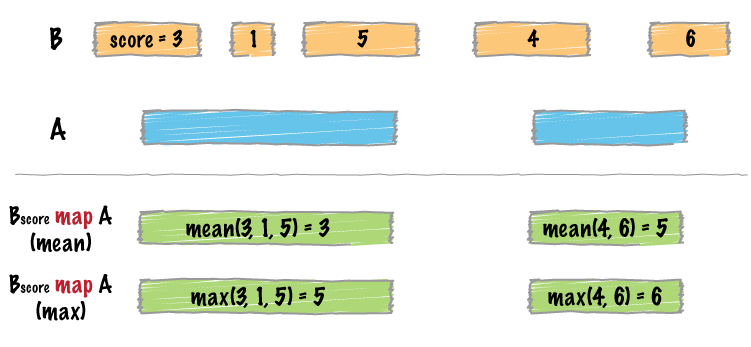
bedtools map requires each input file to be sorted by genome coordinate. For BED files, this can be done with sort -k1,1 -k2,2n. Other sorting criteria are allowed if a genome file (-g) is provides that specifies the expected chromosome order.
The map tool is substantially faster in versions 2.19.0 and later. The plot below demonstrates the increased speed when, for example, counting the number of exome alignments that align to each exon. The bedtools times are compared to the bedops bedmap utility as a point of reference.
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper