What it does
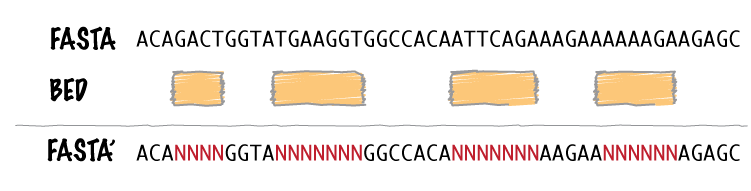
bedtools maskfasta masks sequences in a FASTA file based on intervals defined in a feature file. The headers in the input FASTA file must exactly match the chromosome column in the feature file. This may be useful fro creating your own masked genome file based on custom annotations or for masking all but your target regions when aligning sequence data from a targeted capture experiment.
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper