What it does
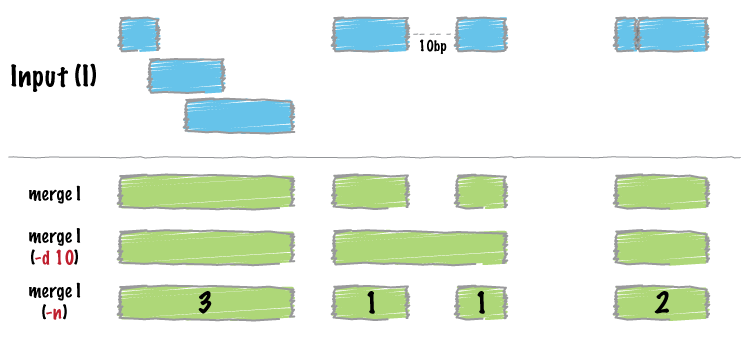
bedtools merge combines overlapping or "book-ended" features in an interval file into a single feature which spans all of the combined features.
bedtools merge requires that you presort your data by chromosome and then by start position.
Default behavior
By default, bedtools merge combines overlapping (by at least 1 bp) and/or bookended intervals into a single, "flattened" or "merged" interval.
$ cat A.bed chr1 100 200 chr1 180 250 chr1 250 500 chr1 501 1000 $ bedtools merge -i A.bed chr1 100 500 chr1 501 1000
-s Enforcing "strandedness"
The -s option will only merge intervals that are overlapping/bookended and are on the same strand.
$ cat A.bed chr1 100 200 a1 1 + chr1 180 250 a2 2 + chr1 250 500 a3 3 - chr1 501 1000 a4 4 + $ bedtools merge -i A.bed -s chr1 100 250 + chr1 501 1000 + chr1 250 500 -
-d Controlling how close two features must be in order to merge
By default, only overlapping or book-ended features are combined into a new feature. However, one can force merge to combine more distant features with the -d option. For example, were one to set -d to 1000, any features that overlap or are within 1000 base pairs of one another will be combined.
$ cat A.bed chr1 100 200 chr1 501 1000 $ bedtools merge -i A.bed chr1 100 200 chr1 501 1000 $ bedtools merge -i A.bed -d 1000 chr1 100 200 1000
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper