What it does
bedtools slop will increase the size of each feature in a feature file by a user-defined number of bases. While something like this could be done with an awk '{OFS="t" print $1,$2-<slop>,$3+<slop>}', bedtools slop will restrict the resizing to the size of the chromosome (i.e. no start < 0 and no end > chromosome size).
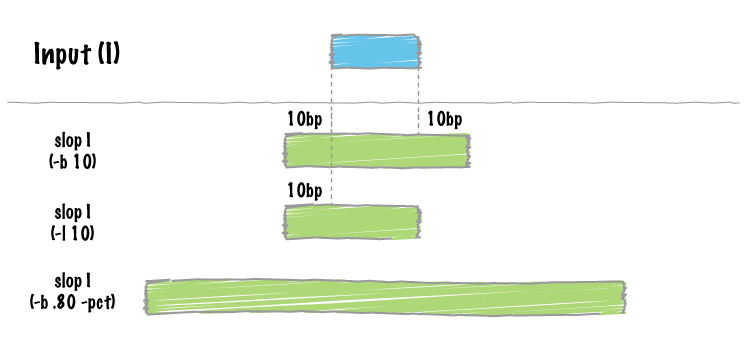
In order to prevent the extension of intervals beyond chromosome boundaries, bedtools slop requires a genome file defining the length of each chromosome or contig.
This tool is part of the bedtools package from the Quinlan laboratory.
Citation
If you use this tool in Galaxy, please cite:
Bjoern A. Gruening (2014), Galaxy wrapper