What it does
This tool samples the given BAM files with paired-end data to estimate the fragment length distribution. Properly paired reads are preferred for computation, i.e., unless a region does not contain any concordant pairs, discordant pairs are ignored.
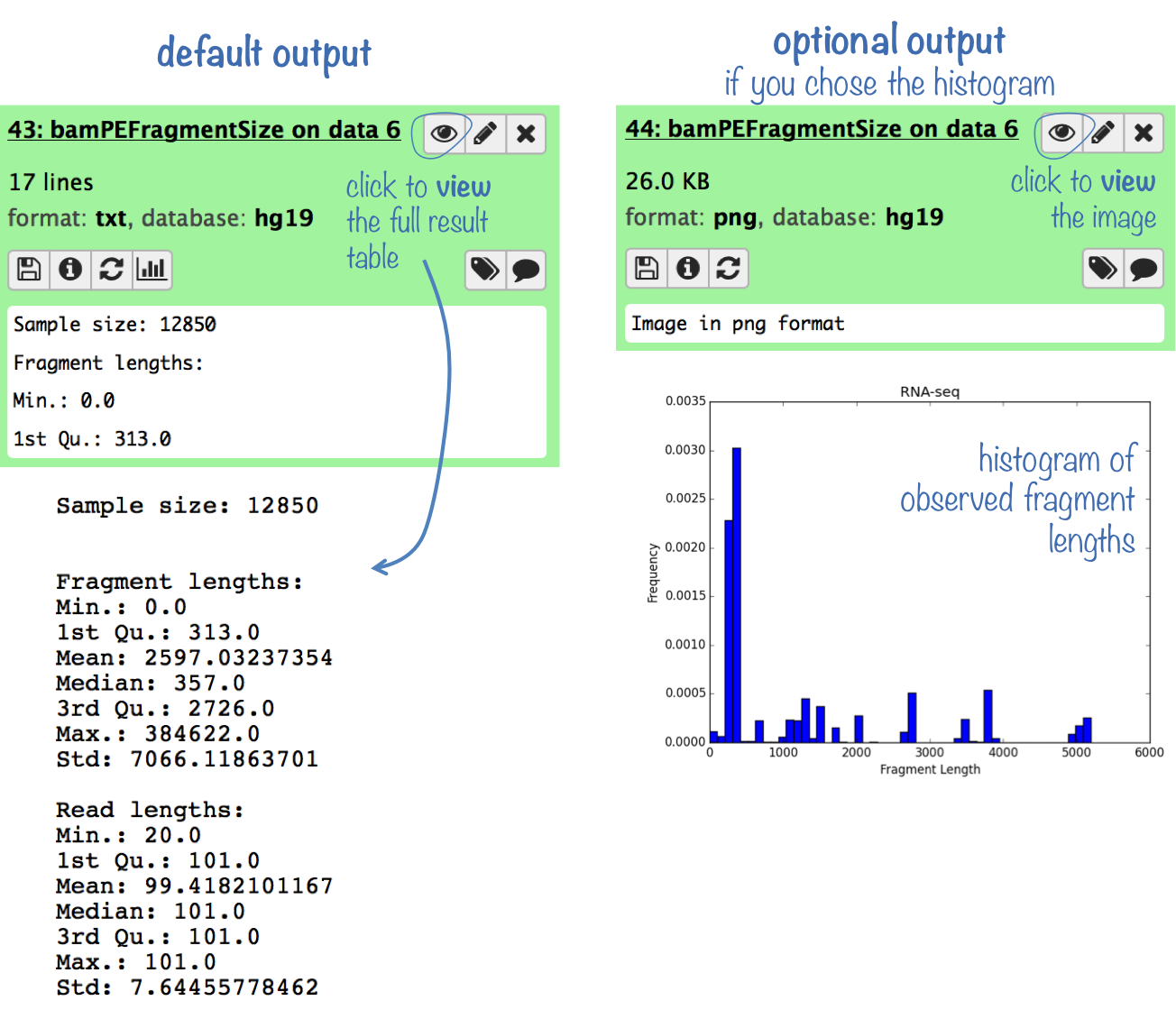
Output
The default output is a simple summary statistic for the observed fragment lengths.
Optionally, you can obtain a histogram of fragment sizes, which will give you a better idea of the distribution of fragment lengths.
If the --table option is specified, the summary statistics are additionally printed in a tabular format:
Frag. Len. Min. Frag. Len. 1st. Qu. Frag. Len. Mean Frag. Len. Median Frag. Len. 3rd Qu. Frag. Len. Max Frag. Len. Std. Read Len. Min. Read Len. 1st. Qu. Read Len. Mean Read Len. Median Read Len. 3rd Qu. Read Len. Max Read Len. Std.
bowtie2 test1.bam 241.0 241.5 244.666666667 242.0 246.5 251.0 4.49691252108 251.0 251.0 251.0 251.0 251.0 251.0 0.0
If the --outRawFragmentLengths option is provided, another history item will be produced, containing the raw data underlying the histogram. It has the following format:
#bamPEFragmentSize Size Occurrences Sample 241 1 bowtie2 test1.bam 242 1 bowtie2 test1.bam 251 1 bowtie2 test1.bam
The "Size" is the fragment (or read, for single-end datasets) size and "Occurrences" are the number of times reads/fragments with that length were observed. For easing downstream processing, the sample name is also included on each row.
For more information on the tools, please visit our help site.
For support or questions please post to Biostars. For bug reports and feature requests please open an issue on github.
This tool is developed by the Bioinformatics and Deep-Sequencing Unit at the Max Planck Institute for Immunobiology and Epigenetics.