Authors Eric Fontanillas created the version 1 of this pipeline. Victor Mataigne developped version 2.
Galaxy integration Julie Baffard and ABiMS TEAM, Roscoff Marine Station
Contact support.abims@sb-roscoff.fr for any questions or concerns about the Galaxy implementation of this tool.Credits : Gildas le Corguillé, Misharl Monsoor
Description
1-Separated mode
Input files are all the orthogroups computed by the AdaptSearch suite; Counts and test are computed on each group separatly. This mode counts occurrences of amino-acids or nucleic acids, according to the sequences type, in each distinct orthogroup. Then, two subgroups of species are set by the user :
- A first group, constitued by species having something in common (an ecological trait, an ecological niche, a particular environmental adaptation)
- A second group, constitued by species sharing the opposite trait (for example, the user can have a first subgroup made with species adapted to high temperatures and a second group made with species adapted to cold temperatures)
Within the groups, the program checks wether the occurrences of each element (amino-acid, nucleic acid, thermostability indice, GC content …) is higher of lower between one species and all the species of the opposite group. Binomial tests are then performed of these counts.
2-Concatenated mode
The input file is the super-alignment obtained by concatenation of all the orthogroups computed by the AdaptSearch suite. This script counts the number of codons, amino acids, and types of amino acids in sequences, as well as the mutation bias from one item to another between 2 sequences. Counts are then compared to empirical p-values, obtained from bootstrapped sequences obtained from a subset of sequences.
In the output files, the pvalues indicate the position of the observed data in a distribution of empirical counts obtained from a resampling of the data. Values above 0.95 indicate a significantly higher count, values under 0.05 a significantly lower count.
The script resamples random pairs of aligned codon to determine what counts can be expected under the hypothesis of an homogenous dataset. Counts are performed on each generated random alignement, thousands of alignments allow to draw a gaussian distribution of the counts. Then the script simply checks whether the observed data are within the 5% lowest or 5% highest values of the distribution.
Input files
If you choose the concatenated method, the input file is the concatenated genes fasta file (in nucleic format) from a previous run of the toolConcatPhyl.
If you choose the separated method, there are two input files : - A dataset collection containing output files from the CDS_Search tool, the one without indels. These files must be in nucleic or proteic format according to the format chosen along with the method. - The concatenated genes fasta file from ConcatPhyl, only used here to retrieve species name.
Parameters
There are parameters only for the "Concatenated" method :
- The list of species for countings, separated by commas and without space (e.g : sp1,sp2,sp3,sp4). You can run the tool on subgroup of species, not only on the total number of species present in the previous tools.
- The list of species for resampling, separated by commas and without space (e.g : sp1,sp2,sp3,sp4). You can run the tool on subgroup of species (at least two species), not only on the total number of species present in the previous tools.
- The number of iterations : the number of alignments that will be generated (effect on the resolution of the gaussian distribution). Shouldn't be lower than 1000 to have a relatively smooth gaussian distribution.
- The number of sampled codons : the number of pairs of codons in each generated alignments (effect on the robustness on the counts performed on this alignement). Shouldn't be lower than 1000 to detect codons with relatively low occurence (<1%).
Outputs
- Many outputs in .csv format , varying according to the chosen method and format (separated, nucleic ...)
- When method = concat : 6 .csv outputs : countings of codons, amino acids, amino acids types, and transitions from amino acid to amino acid and from amino acid type to amino acid type.
- When method = separated and format = nucleic : 4 collections with several .csv files : counts tables and binomial sign tests results for nucleotides and various indices (GC and purine percent ...) .
- When method = separated and format = proteic : 4 collections with several .csv files : counts tables and binomial sign tests results for amino-acids and various indices (thermophilic indices, hydratation, partial specific volume...).
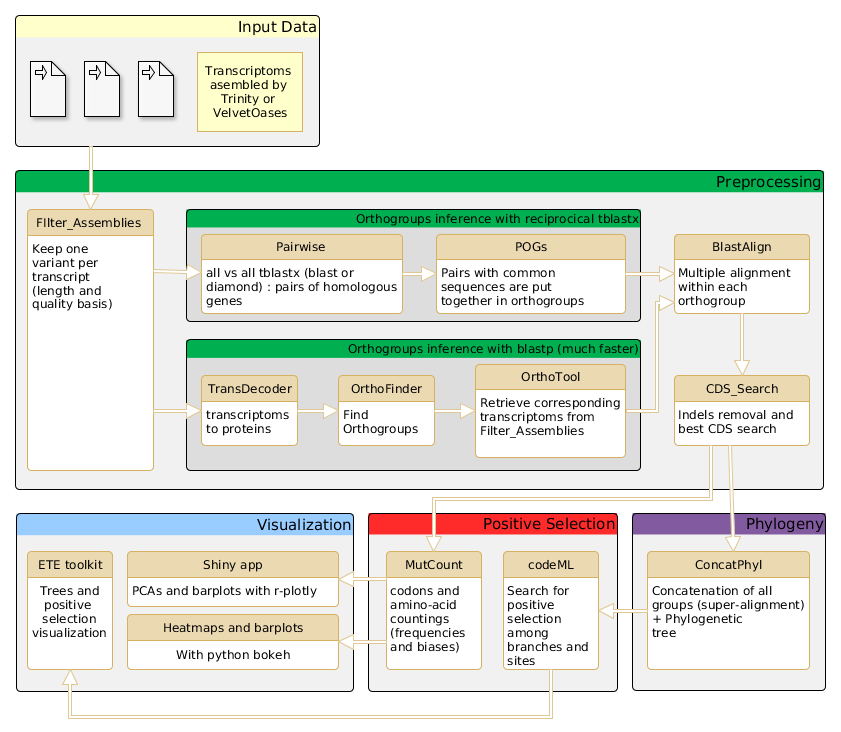
The AdaptSearch Pipeline
Changelog
Version 2.2.0 - 10/07/2018 - Updated separated mode : added a binomial sign test
Version 2.1 - 26/02/2017 - Fully re-written the concat method : fixed mistakes + cleaner code - Splitted output of concatenated method in several csv files. - Bug corrected in output files of separated method.
Version 2.0 - 12/07/2017
- NEW: Replaced the zip between tools by Dataset Collection
- More functional tests
Version 1.0 - 14/04/2017
- Added the tools to the suite
- Added a functional test with planemo
- Planemo test using conda dependencies for python
- Scripts renamed + symlinks to the directory 'scripts'