What it does
Given a BAM file, this tool generates a bigWig or bedGraph file of fragment or read coverages. The way the method works is by first calculating all the number of reads (either extended to match the fragment length or not) that overlap each bin in the genome. Various options are available to normalize the reads: 1) using a given scaling factor 2) RPKM (reads per kilobase per million) : RPKM (per bin) = number of reads per bin / ( number of mapped reads (in millions) * bin length (kb) ). 3) CPM (counts per million) : CPM (per bin) = number of reads per bin / number of mapped reads (in millions). 4) BPM (bins per million) : BPM (per bin) = number of reads per bin / sum of all reads per bin (in millions). 5) RPGC (1x sequencing depth ) : number of reads per bin /(total number of mapped reads * fragment length / effective genome size)
In the case of paired-end mapping, each read mate is treated independently to avoid a bias when a mixture of concordant and discordant pairs is present. This means that each end will be extended to match the fragment length.
See the usage hints below.
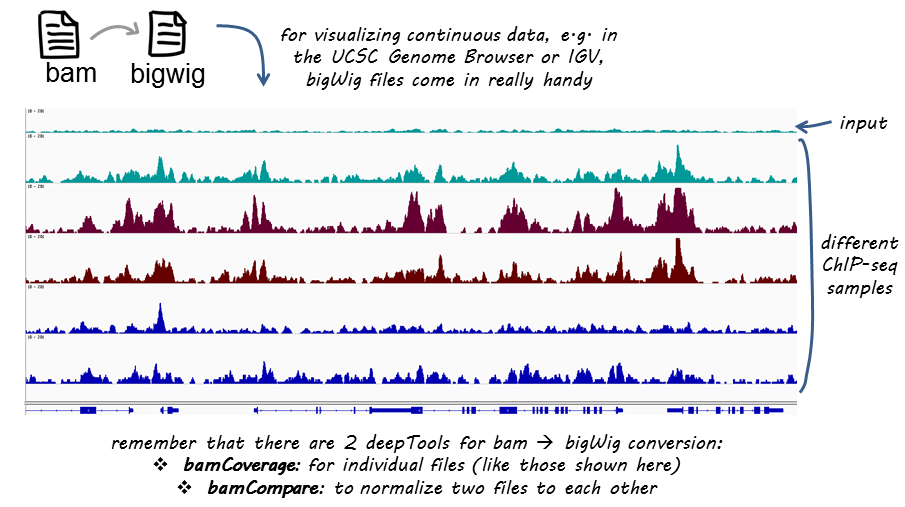
Output
bamCoverage produces a coverage file, either in bigWig or bedGraph format, where for each bin the number of overlapping reads (possibly normalized) is noted.
Like BAM files, bigWig files are compressed, binary files. If you would like to see the coverage values, choose the bedGraph output. For more information on typical NGS file formats, see our Glossary

Usage hints
- A smaller bin size value will result in a higher resolution of the coverage track but also in a larger file size.
- The 1x normalization (RPGC) requires the input of a value for the effective genome size, which is the mappable part of the reference genome. Of course, this value is species-specific.
- It might be useful for some studies to exclude certain chromosomes in order to avoid biases, e.g. chromosome X for many mammals where the males contain a pair of each autosome, but often only a single X chromosome.
- By default, the read length is NOT extended! This is the preferred setting for spliced-read data like RNA-seq, where one usually wants to rely on the detected read locations only. A read extension would neglect potential splice sites in the unmapped part of the fragment. Other data, e.g. ChIP-seq, where fragments are known to map contiuously, should be processed with read extension (--extendReads [INT]).
- For paired-end data, the fragment length is generally defined by the two read mates. The user-provided fragment length is only used as a fallback for singletons or mate reads that map too far apart (with a distance greater than four times the fragment length or if the mates are located on different chromosomes).
WARNING: If you already normalized for GC bias using correctGCbias, you should absolutely NOT set the parameter --ignoreDuplicates!
For more information on the tools, please visit our help site.
For support or questions please post to Biostars. For bug reports and feature requests please open an issue on github.
This tool is developed by the Bioinformatics and Deep-Sequencing Unit at the Max Planck Institute for Immunobiology and Epigenetics.