What it does
This tool compares two bigWig files based on the number of mapped reads. To compare the bigwig files, the genome is partitioned into bins of equal size, then the scores (e.g., number of reads) found in each bigWig file are counted for such bins and, finally, a summary value is reported. This value can be the ratio of the number of reads per bin, the log2 of the ratio, the sum or the difference.
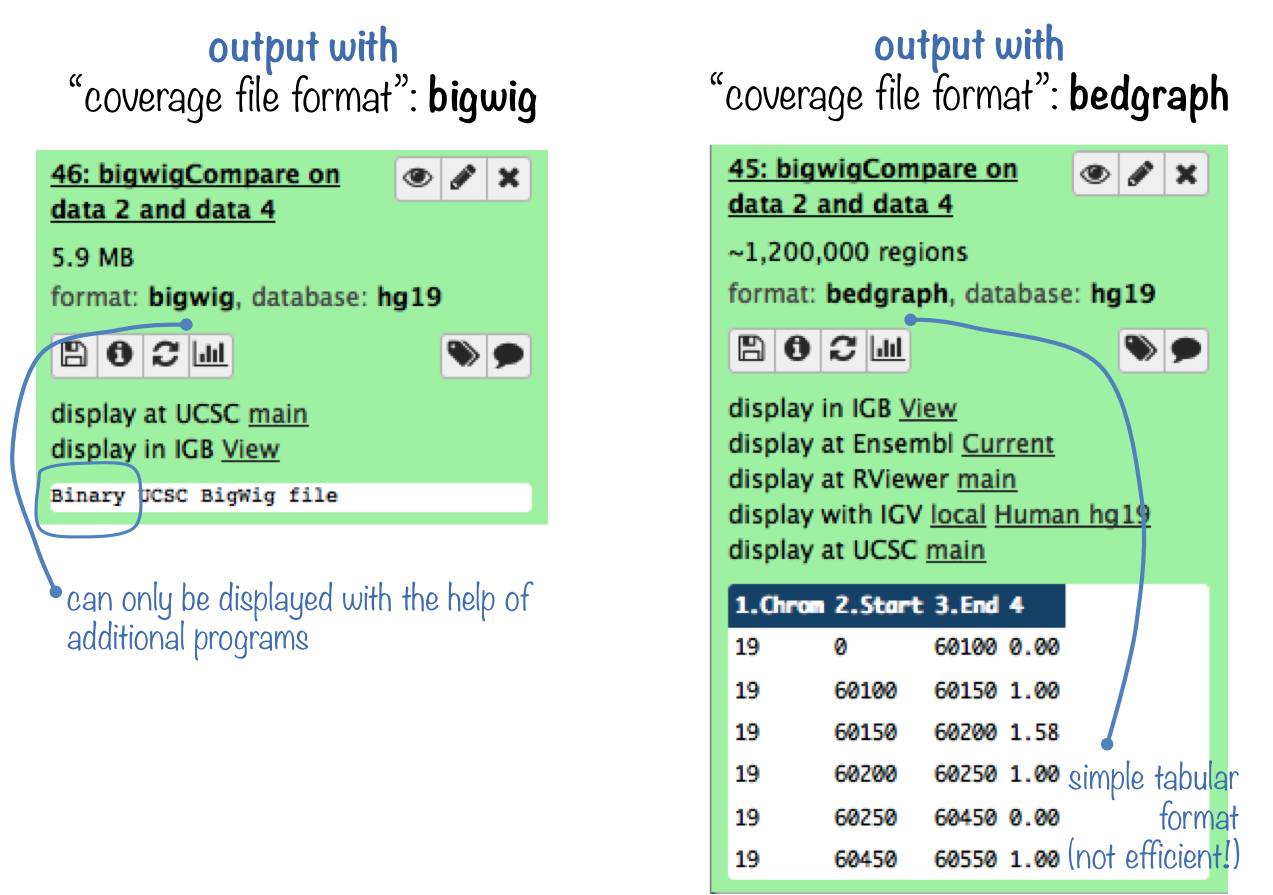
Note that you can actually produce a human-readable bedGraph format instead of the compressed bigWig format if you're interested in having a look at the values yourself.
For more information on the tools, please visit our help site.
For support or questions please post to Biostars. For bug reports and feature requests please open an issue on github.
This tool is developed by the Bioinformatics and Deep-Sequencing Unit at the Max Planck Institute for Immunobiology and Epigenetics.