Authors Eric Fontanillas created the version 1 of this pipeline. Victor Mataigne developped version 2.
Galaxy integration Julie Baffard and ABiMS TEAM, Roscoff Marine Station
Contact support.abims@sb-roscoff.fr for any questions or concerns about the Galaxy implementation of this tool.Credits : Gildas le Corguillé, Misharl Monsoor
Description
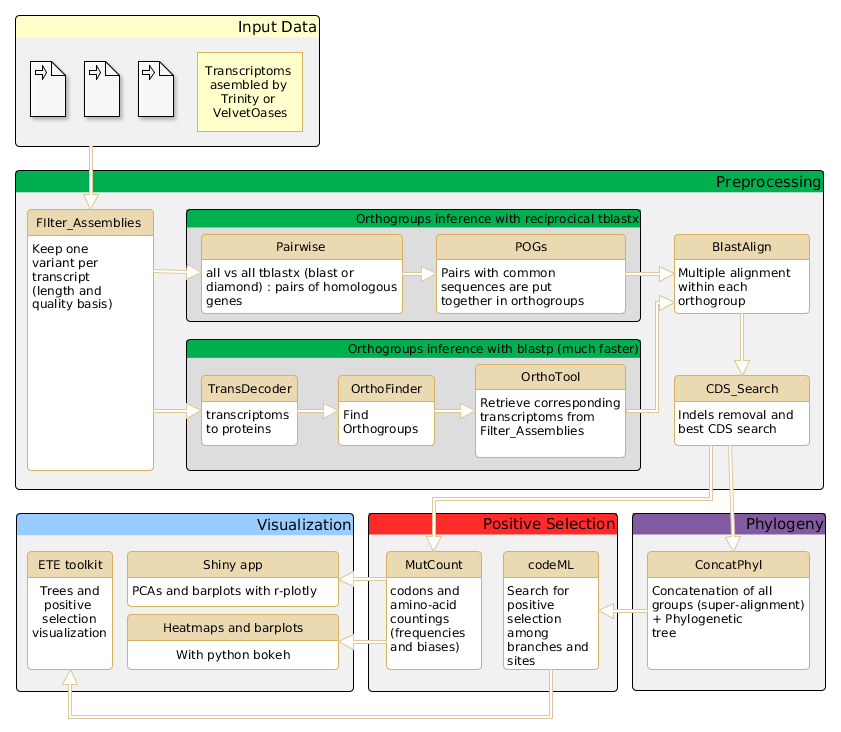
This tool searches for different homologous genes from pairwise comparisons between a set of fasta files (one file per species).
Parameters
- 'Input files' : a collection of fasta files (one file per species)
- 'e_value' : the blast e-value. By default it's 1e-5.
- 'Alignment tool' : choose the sequences alignment tool between tblastx and Diamond. tblastx is more sensitive and Diamond is much faster.
Outputs
- 'Pairwise' : the general output. It gives the information about what the tool has done for each pairwise.
- 'Pairwise_DNA' : the output which contains nucleic sequences (of the pairwise) that are homologous. The sequences are in nucleic format. There are one file per couple of species, and homologous pairs are written by sets of 4 lines :
Exemple for two homologous pairs for the output file for species X/Y :
>Gene_A_species_X_homologous_to_Gene_B_species_Y
Nucleic_sequence_from_filter_assemblies
>Gene_B_species_Y_homologous_to_Gene_A_species_X
Nucleic_sequence_from_filter_assemblies
>Gene_C_species_X_homologous_to_Gene_D_species_Y
Nucleic_sequence_from_filter_assemblies
>Gene_D_species_Y_homologous_to_Gene_C_species_X
Nucleic_sequence_from_filter_assemblies
The AdaptSearch Pipeline
Changelog
Version 2.1 - 03/07/2018
- Add the possibility to use Diamond instead of tblastx
Version 2.0 - 18/04/2017
- NEW: Replace the zip between tools by Dataset Collection
Version 1.0 - 13/04/2017
- TEST: Add functional test with planemo
- IMPROVEMENT: Use conda dependencies for blast, samtools and python