What is does
BAMTools filter is a very powerful utility to perform complex filtering of BAM files. It is based on BAMtools suite of tools by Derek Barnett (https://github.com/pezmaster31/bamtools).
How it works
The tool use logic relies on the three concepts: (1) input BAM, (2) groups, and (3) filters.
Input BAM(s)
The input BAM is self-explanatory. This is the dataset you will be filtering. The tool can accept just one or multiple BAM files. To filter on multiple BAMs just add them by clicking Add new BAM dataset(s) to filter
Conditions and Filters
Conditions for filtering BAM files can be arranged in Groups and Filters. While it can be confusing at first this is what gives ultimate power to this tools. So try to look at the examples we are supplying below.
Example 1. Using a single filter
When filtering on a single condition there is no need to worry about filters and conditions. Just choose a filter from the Select BAM property to filter on: dropdown and enter a value (or click a checkbox for binary filters). For example, for retaining reads with mapping quality of at least 20 one would set the tool interface as shown below:

Example 2. Using multiple filters
Now suppose one needs to extract reads that (1) have mapping quality of at least 20, (2) contain at least 1 mismatch, and (3) are mapping onto forward strand only. To do so we will use three filters as shown below (multiple filters are added to the interface by clicking on the Add new Filter button):
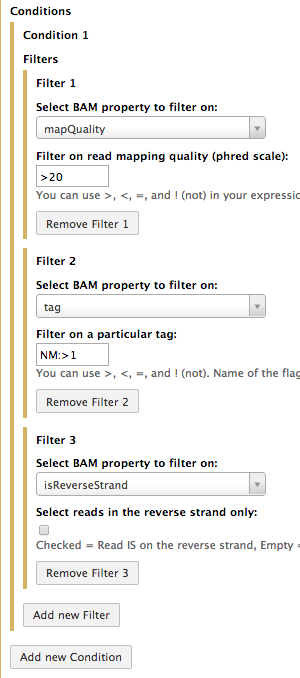
In this case (you can see that the three filters are grouped within a single Condition - Condition 1) the filter too use logical AND to perform filtering. In other words only reads that (1) have mapping quality of at least 20 AND (2) contain at least 1 mismatch AND are mapping onto forward strand will be returned in this example.
Example 3. Complex filtering with multiple conditions
Suppose now you would like to select either reads that (1) have (1.1) no mismatches and (1.2) are on the forward strand OR (2) reads that have (2.1) at least one mismatch and (2.2) are on the reverse strand. In this scenario we have to set up two conditions: (1) and (2) each with two filters: 1.1 and 1.2 as well as 2.1 and 2.2. The following screenshot expalins how this can be done:
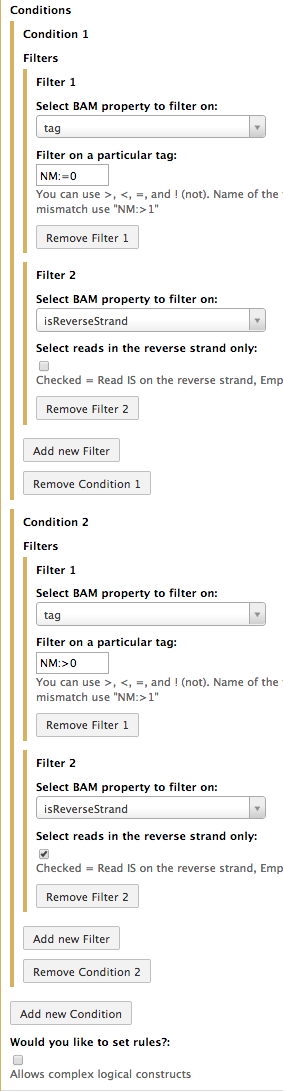
Example 4. Even more complex filtering with Rules
In the above example we have used two conditions (Condition 1 and Condition 2). Using multiple conditions allows to combine them and a variety of ways to enable even more powerful filtering. For example, suppose get all reads that (1) do NOT map to mitochondria and either (2) have mapping quality over 20, or (3) are in properly mapped pairs. The logical rule to enable such filtering will look like this:
!(1) & (2 | 3)
Here, numbers 1, 2, and 3 represent conditions. The following screenshot illustrates how to do this in Galaxy:
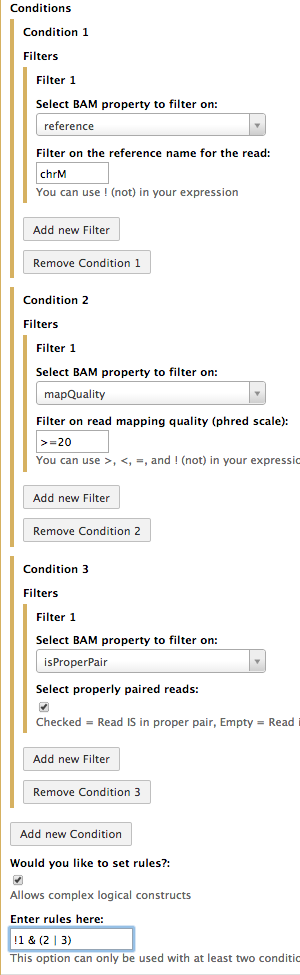
There are three conditions here, each with a single filter. A text entry area that can be opened by clicking on the Would you like to set rules? checkbox enables you to enter a rule. Here numbers correspond to numbers of conditions as they are shown in the interface. E.g., 1 corresponds to condition 1, 2 to condition 2 and so on... In human language this means:
NOT condition 1 AND (condition 2 OR condition 3)
JSON script file
This tool produces two outputs. One of the them is a BAM file containing filtered reads. The other is a JSONified script. It can help you to see how your instructions are sent to BAMTools. For instance, the example 4 looks like this in the JSON form:
{
"filters":
[
{ "id": "1",
"tag":"NM:=0",
"isReverseStrand":"false"
},
{ "id": "2",
"tag":"NM:>0",
"isReverseStrand":"true"
}
]
}
More information
Additional information about BAMtools can be found at https://github.com/pezmaster31/bamtools/wiki