What it does
QUAST = QUality ASsessment Tool. The tool evaluates genome assemblies by computing various metrics.
If you have one or multiple genome assemblies, you can assess their quality with Quast. It works with or without reference genome. If you are new to Quast, start by reading its manual page.
Using Quast without reference
Without reference Quast can calculate a number of assembly related-metrics but cannot provide any information about potential misassemblies, inversions, translocations, etc. Suppose you have three assemblies produced by Unicycler corresponding to three different antibiotic treatments car, pit, and cef (these stand for carbenicillin, piperacillin, and cefsulodin, respectively). Evaluating them without reference will produce the following Quast outputs:
- Quast report in HTML format
- Contig viewer (an HTML file)
- Quast report in Tab-delimited format
- Quast log (a file technical information about Quast tool execution)
The tab delimited Quast report will contain the following information:
Assembly pit_fna cef_fna car_fna # contigs (>= 0 bp) 100 91 94 # contigs (>= 1000 bp) 62 58 61 Total length (>= 0 bp) 6480635 6481216 6480271 Total length (>= 1000 bp) 6466917 6468946 6467103 # contigs 71 66 70 Largest contig 848753 848766 662053 Total length 6473173 6474698 6473810 GC (%) 66.33 66.33 66.33 N50 270269 289027 254671 N75 136321 136321 146521 L50 7 7 8 L75 15 15 16 # N's per 100 kbp 0.00 0.00 0.00
where values are defined as specified in Quast manual
Quast report in HTML format contains graphs in addition to the above metrics, while Contig viewer draws contigs ordered from longest to shortest. This ordering is suitable for comparing only largest contigs or number of contigs longer than a specific threshold. The viewer shows N50 and N75 with color and textual indication. If the reference genome is available or at least approximate genome length is known (see --est-ref-size), NG50 and NG75 are also shown. You can also tone down contigs shorter than a specified threshold using Icarus control panel:
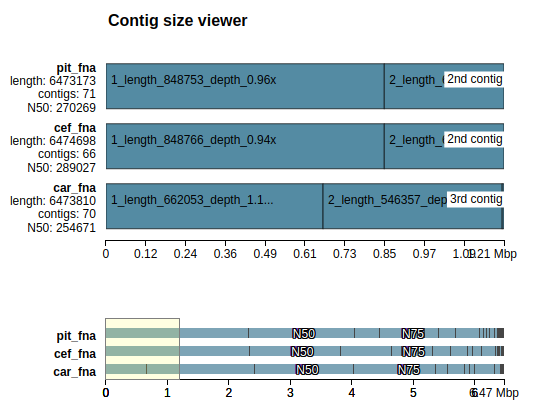
Also see Plot description section of the manual.
Using Quast with reference
Car, pit, and cef are in fact assemblies of Pseudomonas aeruginosa UCBPP-PA14, so we can use its genome as a reference (by supplying a Fasta file containing P. aeruginosa pa14 genome to Reference genome input box). The following outputs will be produced (note the alignment viewer):
- Quast report in HTML format
- Contig viewer (an HTML file)
- Alignment viewer (an HTML file)
- Quast report in Tab-delimited format
- Summary of misassemblies
- Summary of unaligned contigs
- Quast log (a file technical information about Quast tool execution)
With the reference Quast produces a much more comprehensive set of results:
Assembly pit_fna cef_fna car_fna
# contigs (>= 0 bp) 100 91 94
# contigs (>= 1000 bp) 62 58 61
Total length (>= 0 bp) 6480635 6481216 6480271
Total length (>= 1000 bp) 6466917 6468946 6467103
# contigs 71 66 70
Largest contig 848753 848766 662053
Total length 6473173 6474698 6473810
Reference length 6537648 6537648 6537648
GC (%) 66.33 66.33 66.33
Reference GC (%) 66.29 66.29 66.29
N50 270269 289027 254671
NG50 270269 289027 254671
N75 136321 136321 146521
NG75 136321 136321 136321
L50 7 7 8
LG50 7 7 8
L75 15 15 16
LG75 15 15 17
# misassemblies 0 0 0
# misassembled contigs 0 0 0
Misassembled contigs length 0 0 0
# local misassemblies 1 1 2
# unaligned mis. contigs 0 0 0
# unaligned contigs 0 + 0 0 + 0 0 + 0
part part part
Unaligned length 0 0 0
Genome fraction (%) 99.015 99.038 99.025
Duplication ratio 1.000 1.000 1.000
# N's per 100 kbp 0.00 0.00 0.00
# mismatches per 100 kbp 3.82 3.63 3.49
# indels per 100 kbp 1.19 1.13 1.13
Largest alignment 848753 848766 662053
Total aligned length 6473163 6474660 6473792
NA50 270269 289027 254671
NGA50 270269 289027 254671
NA75 136321 136321 146521
NGA75 136321 136321 136321
LA50 7 7 8
LGA50 7 7 8
LA75 15 15 16
LGA75 15 15 17
where, again, values are defined as specified in Quast manual. You can see that this report includes a variety of data that can only be computer against a reference assembly.
Using reference also produces an Alignment viewer:
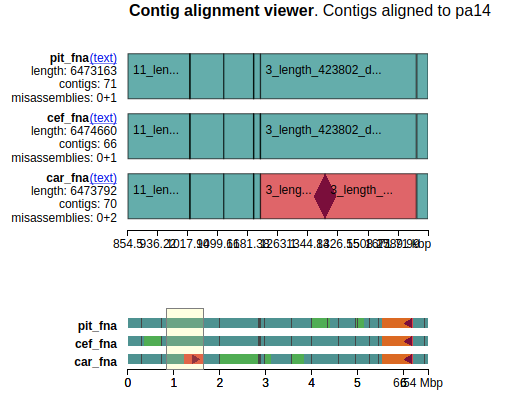
Alignment viewer highlights regions of interest as, in this case, missassemblies that can potentially point to genome rearrangements (see more here).